
(5-氨基-[1,3,4]噻二唑-2-基)-丙酸乙酯的合成工艺改进
2016-08-08 05:43:41陈世强陈宇航
合成化学 2016年7期
陈世强, 朱 飞, 李 鑫, 陈宇航, 陈 向, 余 瑜
(重庆医科大学 药学院,重庆 400016)
·制药技术·
(5-氨基-[1,3,4]噻二唑-2-基)-丙酸乙酯的合成工艺改进
陈世强, 朱飞, 李鑫, 陈宇航, 陈向, 余瑜*
(重庆医科大学 药学院,重庆400016)
摘要:以丁二酸酐为起始原料,经醇解和酰化反应制得丁二酸单乙酯酰氯(3); 3经甲烷磺酸催化与氨基硫脲环合合成了重要药物中间体——(5-氨基-[1,3,4]噻二唑-2-基)-丙酸乙酯,其结构经1H NMR,IR和MS确证。运用正交试验对环合反应条件进行优化。最优反应条件为:3 132 mmol, n(氨基硫脲) ∶n(3) ∶n(甲烷磺酸)=1 ∶3 ∶3,于110 ℃反应3 h,总收率51.3%。
关键词:丁二酸酐; 1,3,4-噻二唑; 丙酸乙酯; 环合; 药物合成; 工艺改进
肿瘤是以细胞异常增殖及其转移为特点的一类疾病,其发病及死亡率一直呈上升趋势,目前已成为严重危害人类健康的一大顽症,因此寻找有效且毒副作用小的抗肿瘤药物对于肿瘤的防治有着重要意义。1,3,4-噻二唑类化合物是重要的药物中间体,具有广泛的生物活性。2-氨基-1,3,4-噻二唑类化合物能抗艾氏腹水癌细胞增殖,对肾癌CAKI-1细胞系和非小细胞肺癌HOP-92细胞系有强烈的抑制作用[1-3];5-硝基芳基-1,3,4-噻二唑类化合物能有效的抑制敏感和耐药性的H.幽门螺旋杆菌的甲硝唑菌株[4];磺酰类1,3,4-噻二唑类化合物表现出高效的抗炎活性,能够抗炎及镇痛且减少脂质过氧化和溃疡等副作用[5];1,3,4-噻二唑类化合物还具有抗惊厥[6]、抗结核[7]、抗寄生虫[8]等生物活性。5-氨基-[1,3,4]噻二唑-2-基)-丙酸乙酯(1)是该类药物的重要中间体,因此,对其合成进行研究具有重要理论意义和实用价值。

Scheme1
关于1的合成方法研究报道较少,经文献调研,酰氯与氨基硫脲直接环合工艺总收率较低[9]。而采用三氯氧磷催化,毒性大、具有刺激性和强腐蚀性[10]。因此,本文以丁二酸酐为原料,经无水乙醇醇解制得丁二酸单乙酯(2),再经氯化亚砜酰化制得丁二酸单乙酯酰氯(3),最后经甲烷磺酸催化与氨基硫脲经环合反应合成1(Scheme1),其结构经1HNMR,IR和MS确证。
1实验部分
1.1仪器与试剂
WC-1型显微熔点仪(温度未校正);AgilentNMRSystems400MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);PerkinElmerSpectrumoneversionB型红外光谱仪(KBr压片);LC-MS-2010A型液质联用仪。
所用试剂均为分析纯。
1.2合成
(1) 2的合成
在圆底烧瓶中加入丁二酸酐60.00g(0.600mol)和无水乙醇60mL(1.029mol),搅拌下回流(100 ℃)反应1h。蒸除过量乙醇得无色透明液体2 86.02g,收率98.2%(97.0%[11])。
(2) 3的合成
在圆底烧瓶中加入2 86.02g(0.589mol)和
氯化亚砜128mL(1.767mol),搅拌下回流(80 ℃)反应2h。蒸除过量氯化亚砜,减压蒸馏(110~120 ℃/4kPa)得无色透明液体3 92.96g,收率95.9%(82.0%[12])。
(3) 1的合成
在圆底烧瓶中加入氨基硫脲4.00g(44mmol)和3 21.80g(132mmol),搅拌下缓慢滴入甲烷磺酸12.80g(0.132mol),滴毕,升温至110 ℃,反应3h。倒入水中,搅拌,用碳酸氢钠调至中性,静置30min,抽滤,滤饼用水洗涤,干燥后用水重结晶得白色针状晶体1 4.82g,收率54.5%,m.p.135~137 ℃;1HNMRδ: 1.263(t, J=7.2Hz, 2H,CH2), 2.793(t, J=6.6Hz, 2H,CH2), 3.206(t, J=7.2Hz, 2H,CH2), 4.144~4.180(m, 2H,OCH2), 5.555(s, 2H,NH2);IRν: 3 294(N—H), 2 979(C—H), 1 733(C=O), 1 640(N=C), 1 348(C—N), 1 174(C—O—C), 1 525(N—H), 1 418(C—H)cm-1;LC-MSm/z: 202{[M+H]+}。
本文以丁二酸酐为原料,经醇解、氯化和环合等3步反应合成1,总收率达51.3%。根据前期预实验发现氨基硫脲与丁二酸单乙酯酰氯配比(A)、氨基硫脲与甲烷磺酸配比(B)、反应温度(C)和时间(D)对合成1影响较大,因此采用正交试验法考察以上4个因素对1收率的影响,选用L9(34)正交表对反应条件进行优化。正交试验因素见表1,结果与分析见表2。

表1 因素与水平
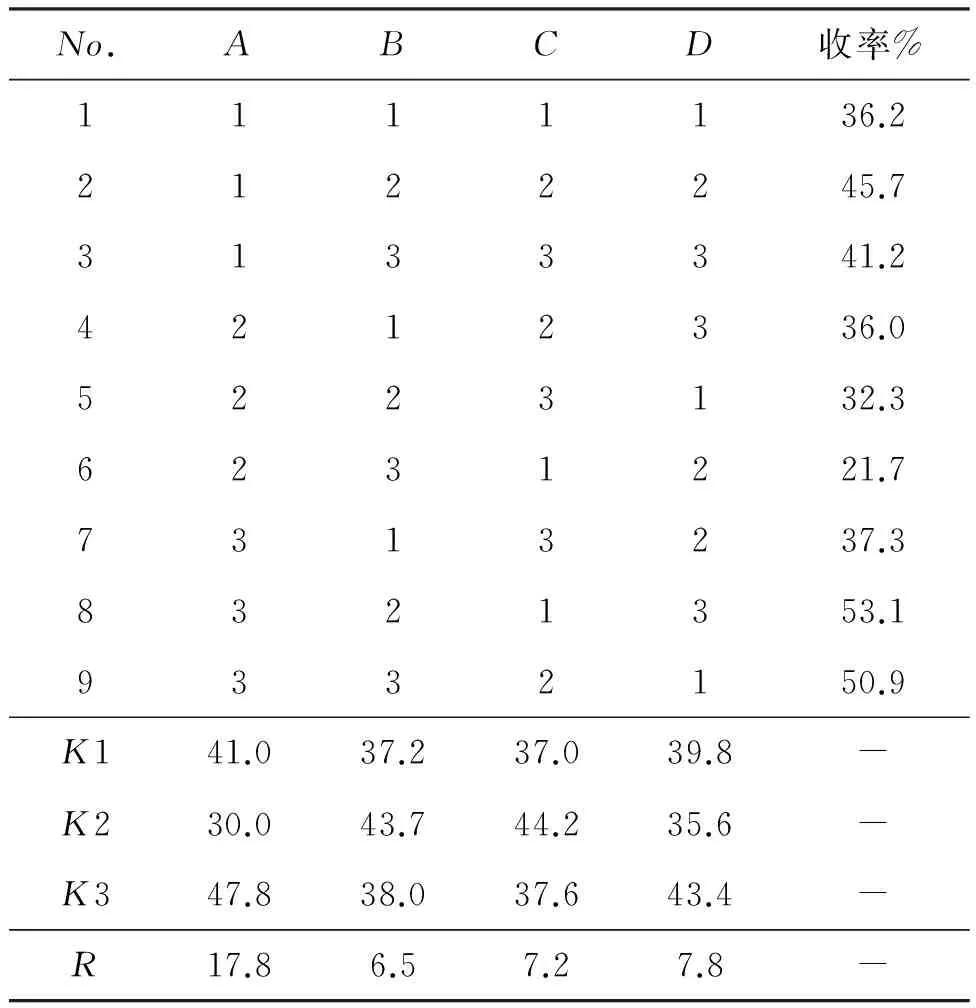
表2 正交实验表及结果
以收率为考察指标,由表2的极差R可知,4种因素对收率影响大小依次为:A>D>C>B,其中因素A对收率的影响最为显著。同时实验结果分析表明A3B2C2D3为最佳实验条件,即n(氨基硫脲) ∶n(酰氯)=1 ∶3, n(氨基硫脲) ∶n(甲烷磺酸)=1 ∶3,于110 ℃反应3h。在该条件下进行3次平行验证实验,收率分别为53.1%, 51.1%和54.5%,平均收率52.9 %。
以丁二酸酐为起始原料合成了重要药物中间体——(5-氨基-[1,3,4]噻二唑-2-基)-丙酸乙酯。对环合的反应条件进行了正交试验优化,在最佳实验条件下,总收率51.3 %。该合成方法使用不挥发、安全性好的甲烷磺酸代替毒性强的三氯氧磷,具有操作简便、成本低廉、绿色环保和后处理安全等优点,具有潜在的工业化前景。
参考文献
[1]GuanP,SunFE,HouXB, et al.Design,synthesisandpreliminarybioactivitystudiesof1,3,4-thiadiazolehydroxamicacidderivativesasnovelhistonedeacetylaseinhibitors[J].BioorgMedChem,2012,20(12):3865-3872.
[2]MalleshappaNN,HarunMP,SaritaK, et al. 2,6-Disubstitutedimidazo[2,1-b][1,3,4]thiadiazoles:Searchforanticanceragent[J].EuropeanJournalofMedicinalChemistry,2012,56:56-69.
[3]AhadAM,ZhouLL,MashEA, et al.DevelopmentofsulfonamideAKTPHdomaininhibitors[J].BioorgMedChem,2011,19(6):2046-2054.
[4]ForoumadiA,RinehA,EmamiS, et al.Synthesisandanti-Helicobacterpyloriactivityof5-(nitroaryl)-1,3,4-thiadiazoleswithcertainsulfurcontainingalkylsidechain[J].BioorgMedChemLett,2008,18(11):3315-3320.
[5]AmirM,KumarH,JavedSA.Non-carboxylicanaloguesofnaproxen:Design,synthesis,andpharmacologicalevaluationofsome1,3,4-oxadiazolthiadiazoleand1,2,4-triazolederivatives[J].ArchPharm,2007,340(11):577-585.
[6]ChapleoCB,MyersM,MyersPL, et al.Substituted1,3,4-thiadiazoleswithanticonvulsantactivityhydrazines[J].JMedChem,1986,29(11):2273-2280.
[7]ForoumadiA,MirzaeiM,ShafieeA.Antituberculosisagents-IIEvaluationofin vitroantituberculosisactivityandcytotoxicityofsome2-(1-methyl-5-nitro-2-imidazolyl)-1,3,4-thiadiazolederivatives[J].Farmaco,2001,56(8):621-623.
[8]BodaC,EnangaB,DumetH, et al.Plasmakineticsandefficacyoforalmegazoltreatmentintrypanosomabruceibrucei-infectedsheep[J].VetParasitol,2004,121(3-4):213-223.
[9]MasakiO.Studieson1,3,4-thiodiazolederivatives.V.Synthesisofcarboxylicacidderivatives[J].JAmChemSoc,1952,72(12):1536-1537.
[10]ClarkJH,EnglishJP,WinnekPS, et al.StudiesinchemotherapyXIIsomesulfanilamidoheterocycles[J].JAmChemSoc,1946,68(1):96-99.
[11]EisenführA,AroraPS,SengleG, et al.ARibozymewithmichaelaseactivity:Synthesisofthesubstrateprecursors[J].BioorganicandMedicinalChemistry,2003,11(2):235-249.
[12]ClementJA,MohanakrishnanAK.Synthesisandcharacterizationofnaphth-annelatedthiopheneanalogs[J].Tetrahedron,2010,66(13):2340-2350.
收稿日期:2016-04-14
基金项目:国家自然科学基金资助项目(81172097, 30371632)
作者简介:陈世强(1990-),男,汉族,重庆人,本科,主要从事药物化学研究。 E-mail: chenshiqiang1990@126.com 通信联系人: 余瑜,教授, E-mail: yuyu3519@163.com
中图分类号:O621.3; R914.5
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.07.16106
ProcessImprovementontheSynthesisof(5-Amino-[1,3,4]thiadiazol-2-yl)-propionicAcidEthylEster
CHENShi-qiang,ZHUFei,LIXin,CHENYu-hang,CHENXiang,YUYu*
(CollegeofPharmaceuticalSciences,ChongqingMedicalUniversity,Chongqing400016,China)
Abstract:Succinic acid monoethyl ester chloride(3) was prepared by alcoholysis, acetylation reaction from succinic anhydride. The important intermediate, (5-amino-[1,3,4]thiadiazol-2-yl)-propionic acid ethyl ester, was synthesized by cyclization of 3 with thiosemicarbazide catalyzed by methane sulfonic acid. The structure was confirmed by1H NMR, IR and MS. The reaction conditions for cyclizating reaction was investigated by the orthogonal design. The results showed that the optimal reaction conditions were as followed: 3 132 mmol, n(thiosemicarbazide) ∶n(3) ∶n(methane sulfonic acid)=1 ∶3 ∶3, reaction at 110 ℃ for 3 h, the total yield was 51.3%.
Keywords:succinic anhydride; 1,3,4-thiadiazole; propionic acid ethyl ester; cyclization; drug synthesis; process improvement
猜你喜欢
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19 08:38:52
兴义民族师范学院学报(2018年5期)2018-12-18 03:16:54
广州城市职业学院学报(2016年2期)2016-07-25 07:39:30
现代食品(2016年24期)2016-04-28 08:12:06
化工进展(2015年3期)2015-11-11 09:07:41
中国生化药物杂志(2015年4期)2015-07-07 12:05:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
湖南师范大学自然科学学报(2015年2期)2015-02-27 14:50:13
应用化工(2014年3期)2014-08-16 13:23:50
中国药业(2014年21期)2014-05-26 08:56:50
