
Kartagener综合征3例并文献复习*
2016-07-31 17:28:55周小青孔晋亮蔡双启
重庆医学 2016年34期
周小青,王 可,孔晋亮,蔡双启,叶 萍
(广西医科大学第一附属医院呼吸疾病研究所,南宁 530021)
·短篇及病例报道·
Kartagener综合征3例并文献复习*
周小青,王 可,孔晋亮△,蔡双启,叶 萍
(广西医科大学第一附属医院呼吸疾病研究所,南宁 530021)
Kartagener 综合征(Kartagener′s Syndrome,KS)又称支气管扩张-鼻窦炎-内脏转位综合征,是一种罕见的先天性常染色体隐性遗传疾病。早期正确诊断对KS患者的预后非常重要,但因本病少见,临床医生对其认识不足而易误诊、漏诊。为提高临床医生对本病的认识,现对本科2006年1月至2016年3月收治的3例患者资料进行分析,总结其临床特点并复习相关文献资料如下。
1 临床资料
病例1,患者,女,77岁5个月,因“反复咳嗽、咳痰8余年,加重伴呼吸困难半年余,再发18 d”入院。患者适龄结婚,育有5子1女,否认相似家族病史及遗传病史。查体:双上肺可闻及干性啰音,上下肺可闻及少许湿性啰音。心尖搏动位于右侧第五肋间锁骨中线内0.5 cm。鼻咽部+胸部CT提示:(1)鼻咽部CT平扫未见明显异常;(2)左侧上颌窦炎症;(3)两肺Ⅲ型肺结核;(4)右弓右降。腹部B超提示内脏反位,肝脏、胆囊位于左上腹,脾脏位于右上腹;肝多发囊肿。心脏B超:(1)先天性镜像右位心;(2)内脏反位;(3)左室顺应性降低,左室收缩功能测定在正常范围。入院诊断:(1)支气管扩张并感染;(2)内脏反位。入院后予抗感染(哌拉西林钠他唑巴坦钠+左氧氟沙星)、止咳化痰、护肝护胃、纤支镜肺泡灌洗及支持对症治疗后,症状改善,病情好转出院。出院诊断:Kartagener 综合征。见图1。
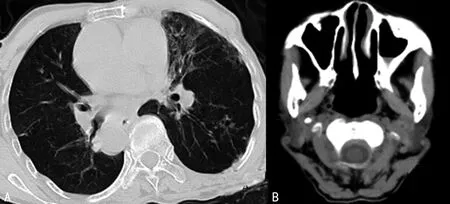
A:胸部;B:鼻窦。
图1 病例1患者胸部、鼻窦CT
病例2,患者,女,28岁6个月,因“反复咳嗽、咳痰20年,加重1周”入院。患者自诉20多年前开始出现反复咳嗽,起病初咳白痰为主,量较少,此后患者咳嗽、咳痰逐年增多,以咳黄脓痰为主,近2年患者咳痰每天20~100 mL。曾在当地医院诊断“肺炎、支气管扩张、右位心”,给予抗感染等治疗后可好转,但症状反复。1周前患者受凉后咳嗽、咳痰症状加重,咳黄痰,门诊以“支气管扩张”收入本科室。适龄结婚,配偶体健,结婚6年,不孕。否认父母近亲结婚,否认相似家族病史及遗传病史。查体:左下肺闻及少量干鸣音,双下肺闻及湿性啰音。心尖搏动位于右侧第五肋间锁骨中线内0.5 cm。胸部X线片示:(1)左肺炎症,待除外支气管扩张或肺囊肿;(2)镜面右位心。胸部CT提示:(1)两肺炎症;(2)左肺上叶下舌段支气管扩张并肺膨胀不全;(3)镜面右位心。鼻窦CT示双侧上颌窦、筛窦、蝶窦炎症,双侧下鼻甲肥大。入院后予抗感染(哌拉西林他唑巴坦+克林霉素),止咳祛痰,调节免疫功能等对症支持治疗后好转,复查胸部CT示肺炎明显吸收,予出院。出院诊断:Kartagener 综合征。见图2。
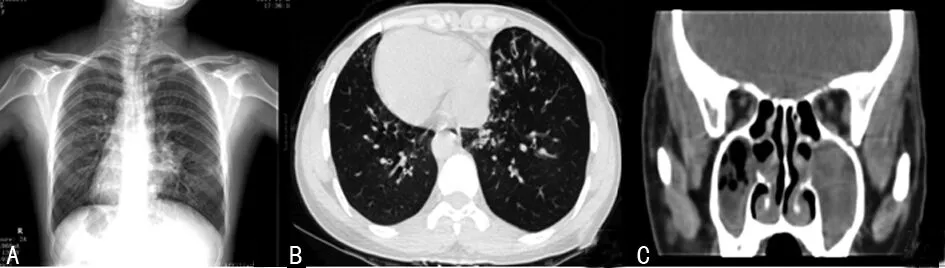
A:胸部X线片;B、C:CT。
图2 病例2患者胸部X线、CT检查
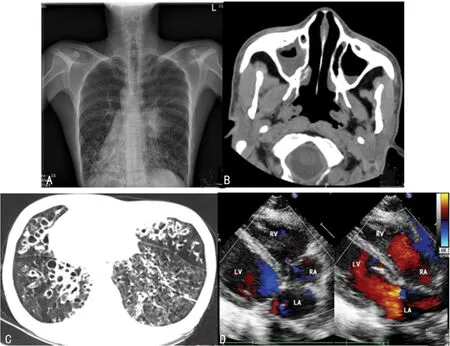
A:胸部X线片;B、C:胸部CT;D:心脏彩色多普勒超声。
图3 病例3患者胸部X线、CT、心脏彩色多普勒超声检查
病例3,患者,男,31岁9个月,因“反复咳嗽、咳痰10年,再发伴发热3 d”入院。患者自幼体弱,极易“感冒”。10年前无明显诱因下出现阵发性连声咳,多为咳白色黏痰,偶有黄色脓痰,受凉后及秋冬季加重,至当地医院抗感染治疗(具体不详)后上症可好转,但10年来反复发作。3 d前患者受凉后出现咳嗽、咳痰再发,咳嗽性质及程度同前,咳大量黄色脓痰,每天约20 mL,伴发热,最高体温可达38.0 ℃,至本院门诊就诊,血常规示白细胞计数 13.07×109/L,血小板计数 398.40×109/L,中性粒细胞绝对值 9.52×109/L,中性粒细胞百分比 0.729。红细胞沉降率68 mm。胸部X线片示:(1)两下肺炎症;(2)右位心。门诊以“肺炎”收入本科室。发现右位心20余年,具体不详。适龄结婚,配偶体健,不孕,领养1女。父母均体健,否认父母近亲结婚,妹妹有鼻窦炎病史,具体不详。对姜和乙醇过敏。查体:双肺呼吸音粗,双下肺可闻及少量湿啰音,未闻及干啰音及胸膜摩擦音。心尖博动位于右侧第5肋间锁骨中线内0.5 cm。双手可见杵状指。入院后完善相关检查,予抗感染(哌拉西林钠舒巴坦钠+盐酸克林霉素)、止咳化痰、护肝护胃、纤支镜肺泡灌洗及对症支持治疗后,症状改善,病情好转出院。见图3。
2 讨 论
2.1 流行病学 1904年由Siewert报道;1933年Kartagener对22个家族中的48例患者进行了详尽的描述而命名;1981年,Rossmon等提出将该综合征命名为原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)。 PCD包括Kartagener综合征又称支气管扩张-鼻窦炎-内脏转危综合征(KS)、不动纤毛综合征和纤毛运动方向缺陷,预计患病率为1/4 000~1/40 000不等。O′Callaghan等[1]对在英国的亚洲人群进行调查时发现,患者的父母为堂兄妹/表兄妹近亲结婚的患病率高达4.41%。KS为PCD的特殊亚型,伴内脏转位,男女患病率无明显差别,占PCD的56%[2]。
2.2 发病机制 1975年Comner和其同事通过观察2例患者的呼吸道黏膜上皮,提出KS的发病机制为纤毛运动障碍[3]。扫描电子显微镜下可观察到KS患者有不同的纤毛超微结构缺陷,包括放射性轮辐丝的缺失、微管的错位或内、外动力蛋白臂的缺失异常等[4-5]。纤毛广泛存在鼻咽部、中耳、鼻窦、及气管至终末呼吸道,其病理基础是由于呼吸道纤毛活动异常削弱黏液清除能力,导致分泌物潴留,细菌和病毒滋生,从而导致上、下呼吸道反复或持续感染,发生不可逆支气管扩张和鼻旁窦炎[6]。同时,纤毛还广泛存在输卵管、输精管、精子鞭毛、脑、脊髓室管膜等组织器官中;胚胎发育过程中纤毛运动障碍导致胎儿内脏随机转位,有一半表现为内脏反位;女性由于输卵管内纤毛运动障碍,影响卵子排出、卵子与精子结合而出现异位妊娠,甚至不孕;男性则因精子尾部鞭毛结构异常,引起精子活动能力不足导致不育。本文病例1有生育能力,病例2和病例3无生育能力。
随着对KS/PCD研究的深入和二代测序技术(NGS)的发展,目前发现存在近30种基因变异,它们包括DNAI1、DNAH5、SPAG1、CCDC40、CCDC39、CCDC114、CCDC103、DNAH11、DNAI2、DNAL1、NME8、RSPH9、RSPH4A、RSPH1、OFD1、RPGR等;其中DNAH5突变约占外动力蛋白臂缺陷患者的50%,DNAI1约占10%;SPAG1变异约占内外动力蛋白臂同时缺陷患者的10%[7]。有研究应用全外显子组测序1例患者发现c.8030G>A(p.R2677Q)突变,位于基因DNAH5[8];另一研究对DNAH1序列分析发现一种纯外显子20的错配变异(g.52387629A>C;c.3460A>C;p.Lys1154Gln)[9]。
2.3 临床表现 主要表现为反复发生的慢性鼻炎、鼻窦炎、慢性支气管炎、支气管扩张及反复发生的肺炎;也可伴发其他多系统疾病,如多囊肝、多囊肾、视网膜病变、脑积水、胆道闭锁、慢性中耳炎、传导性耳聋、男性不育、女性生育能力下降、宫外孕倾向、严重的慢性头痛等。症状表现多种多样,严重程度各不相同,包括咳嗽、咳(脓)痰、咯血、鼻腔分泌物增多、呼吸困难等,随年龄增长而逐步加重;呼吸道感染时肺部有干湿性啰音;可有发绀、杵状指等。本文3例患者均出现反复呼吸道感染,慢性咳嗽、咳痰病史,2例有不育不孕病史,1例呼吸困难,1例肝多发囊肿,查体3例均有肺部干湿啰音,1例患者有杵状指。
2.4 诊断 诊断标准:(1)右位心或全内脏转位;(2)支气管扩张症;(3)鼻窦炎。KS根据支气管扩张、内脏转位和鼻窦炎三联征的发生情况分为完全型和不完全型,三联征均发属完全型KS,如仅有支气管扩张和内脏转位为不完全型KS,右位心为诊断的必备条件。本文报道3例均为完全型KS。根据国外文献报道,有下列情况时应考虑本病的可能:(1)内脏反转伴呼吸系统或鼻部症状;(2)不明原因新生儿呼吸窘迫症;(3)兄弟姐妹为原发性纤毛运动障碍(PCD)患者,特别是伴相应临床症状时;(4)长期咳嗽、咳痰,可从幼年开始出现而没有引起父母重视;(5)如果考虑囊性纤维化,亦可考虑PCD,特别是伴有鼻炎,鼻窦炎或中耳炎的患者;(6)原因不明的支气管扩张症;(7)浆液性中耳炎伴上、下呼吸道症状;(8)心脏病伴内脏异位,且怀疑有呼吸道,鼻腔或耳部疾病;如果患者有PCD家族史,则考虑该病的门槛应该降低[7]。
2.5 治疗 目前尚无能使纤毛功能恢复的药物,现阶段仍以对症支持治疗、预防反复感染为主。急性期主要采取化痰、排痰、体位引流、止血及根据药敏试验选择敏感的抗菌药物;缓解期应加强锻炼,营养支持以增强身体免疫力,可适当使用免疫调节剂、接种疫苗,以减少感染概率。呼吸系统的良好管理可提高患者肺功能,延缓疾病进展。Ebner等研究表明,钙离子载体对Kartagener综合征患者有一定疗效[10]。本文3例患者经过吸氧、积极抗感染、止咳祛痰、护肝护胃、经纤维支气管镜吸引气管内分泌物、肺泡灌洗、调节免疫功能等对症支持治疗后,症状均改善,病情好转出院。
2.6 预后 本病的预后与支气管扩张程度相关,早期诊断和有效干预可延缓本病的发展[4]。大多数患者预后良好,但如反复长期呼吸道感染者最终可发展成呼吸功能不全或心力衰竭。
KS是一种罕见的遗传性疾病,其诊断并不困难,但是由于临床医生对该病的认识不足,常会误诊、漏诊,从而延误治疗。希望通过本研究,使临床医生对该病有更深的认识,做到早发现、早诊断、早治疗,从而改善患者的预后,延长预期寿命。
[1]O′Callaghan CP,Chetcuti EM.High prevalence of primary ciliary dyskinesia in a British Asian population[J].Arch Dis Child,2010,95(1):51-52.
[2]Kennedy MP,Noone PG,Leigh MW,et al.High-resolution CT of patients with primary ciliary dyskinesia[J].AJR Am J Roentgenol,2007,188(5):1232-1238.
[3]Camner P,Mossberg B,Afzelius BA.Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects[J].Am Rev Respir Dis,1975,112(6):807-809.
[4]Ciancio N,de Santi MM,Campisi R,et al.Kartagener′s syndrome:review of a case series[J].Multidiscip Respir Med,2015,10(1):18.
[5]Tanaka K,Sutani A,Uchida Y,et al.Ciliary ultrastructure in two sisters with Kartagener′s syndrome[J].Med Mol Morphol,2007,40(1):34-39.
[6]Sleigh MA, Blake JR,Liron N.The propulsion of mucus by cilia[J].Am Rev Respir Dis,1988,137(3):726-741.
[7]Lucas JS,Burgess A, Mitchison HM,et al.Diagnosis and management of primary ciliary dyskinesia[J].Arch Dis Child,2014,99(9):850-856.
[8]张静,白银,尤少华,等.Kartagener综合征合并分泌性中耳炎患者的基因诊断[J].中华耳科学杂志,2014,12(1) :41-44.
[9]Imtiaz F,Allam R,Ramzan K,et al.Variation in DNAH1 may contribute to primary ciliary dyskinesia[J].BMC Med Genet,2015(16):14.
[10]Ebner T, Maurer M, Oppelt P,et al.Healthy twin live-birth after ionophore treatment in a case of theophylline-resistant Kartagener syndrome[J].J Assist Reprod Genet,2015,32(6):873-877.
10.3969/j.issn.1671-8348.2016.34.047
国家自然科学基金资助项目(81260663);广西自然科学基金项目(2013GXNSFAA019166)。 作者简介:周小青(1982-),在读硕士,主要研究领域为肺部感染性疾病的基础研究。△
,E-mail:kjl071@163.com。
R596.1
C
1671-8348(2016)34-4890-03
2016-05-08
2016-08-06)
猜你喜欢
实用医院临床杂志(2023年4期)2023-07-31 01:57:48
自然杂志(2022年3期)2022-08-18 03:00:06
散文诗世界(2019年6期)2019-09-10 07:22:44
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:56
意林·全彩Color(2019年7期)2019-08-13 00:53:46
中华皮肤科杂志(2019年5期)2019-06-24 06:32:06
实用心电学杂志(2018年5期)2018-10-24 06:49:18
中国介入影像与治疗学(2017年1期)2017-02-21 09:07:10
西南医科大学学报(2015年1期)2015-08-22 13:01:56
天津医药(2011年1期)2011-03-16 21:48:53
