
过渡金属氧化物催化腈水合生成酰胺的研究进展
2016-07-26 09:52赵晓甫张月成张宏宇赵继全河北工业大学化工学院天津300130
化工进展 2016年7期
赵晓甫,张月成,张宏宇,赵继全(河北工业大学化工学院,天津300130)
综述与专论
过渡金属氧化物催化腈水合生成酰胺的研究进展
赵晓甫,张月成,张宏宇,赵继全
(河北工业大学化工学院,天津300130)
摘要:腈水合生成酰胺具有原子经济性高以及无其他副产物生成的优点。通常腈水合生成酰胺是在强酸或强碱催化下进行的,但其具有腈过度水解生成羧酸以及需要中和催化剂生成盐等缺点。采用过渡金属氧化物如氧化锰(MnO2)、氧化镍(NiO)、氧化铈(CeO2)以及氢氧化钌[Ru(OH)x]为催化剂替代强酸或强碱可克服上述缺点。本文总结了上述过渡金属氧化物为催化剂催化腈水合生成酰胺反应的进展,从中可以看到腈水合生成酰胺反应依赖于催化剂的种类、制备方法以及腈的结构。对每种催化剂的制备方法、使用范围、优缺点进行了分析。对各种催化剂催化腈水合生成酰胺反应的可能机理进行讨论。根据以上讨论,预期此类催化剂将向复合型和负载型方向发展。
关键词:水合;腈;酰胺;过渡金属氧化物;二氧化锰;氧化镍;二氧化铈;氢氧化钌
酰胺是一类非常重要的有机化合物,在药物合成、化工生产以及生物学领域有着广泛的应用。如酰胺可用作溶剂、肽和蛋白质合成的中间体、高分子单体、彩色墨水的颜料、洗涤剂以及润滑剂等[1-4]。传统的酰胺合成方法是由羧酸以及更高反应活性的羧酸衍生物酰氯、酸酐以及酯与有机胺或氨气在适当温度下通过缩合反应得到[5]。该方法原子经济性低,分离困难,伴生的副产物对环境造成污染,因此,改进酰胺的合成方法是近年来有机合成、制药和化工生产领域挑战性和迫切的课题之一[6]。基于此,化学家对酰胺的合成进行了广泛研究,发现和发展了多种由各种原料获得酰胺的方法[7-9]。在众多的合成酰胺的方法中,由腈水合获得酰胺具有最高的原子经济性[反应式(1)]。而腈的合成可以由醇的催化氨化-脱氢[10-13]以及醇或甲基芳烃的氧化氨化[14-16]得到。理论上前者的副产物为氢气和水,后者的副反物只有水,因此,也具有较高的原子经济性及对环境友好的优点。综合而言,由醇和氨最终得到酰胺,无论是从经济的角度还是从环保的角度考虑,都具有其他传统方法无可比拟的优越性。
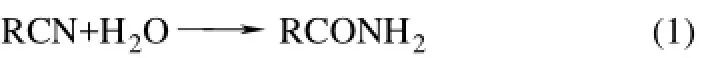
但是,由腈水合生成酰胺需要在催化剂催化下才能顺利进行。传统的催化剂是强酸和强碱,而且反应条件苛刻,不适合结构共存其他活性基团的腈类分子的水合。此外,当用强碱为催化剂时,会发生酰胺的过度水解生成相应的羧酸,因为酰胺水解生成酰胺相较于腈水合生成酰胺在动力学上是有利的。尽管在强酸催化下腈水合反应可停留在酰胺阶段,但必须严格控制反应条件,包括反应温度以及腈与水要严格等化学剂量,否则会发生聚合反应。实际上,无论是采用酸催化还是碱催化,在生产工艺中都需要碱或酸中和,而中和过程形成的盐不利于产物的分离纯化并对环境产生污染。为克服强酸、强碱催化腈水合生成酰胺反应的缺点,人们先后开发了多种可用于腈水合生成酰胺的均相、多相化体系。其中基于过渡金属配合物的均相催化体系具有催化活性和对目标化合物酰胺选择性高的优点,但也存在催化剂价格昂贵、难于与产物分离因而造成不能循环使用的缺点。相关文献已对此类催化剂进行了全面的综述[9,17]。另外,一些基于过渡金属氧化物的多相催化剂在腈水合生成酰胺反应中显示良好的催化性能。研究发现,此类催化剂的催化性能的优劣依赖于过渡金属的种类以及氧化物的制备方法。从反应原理的角度看,此类催化剂既不同于传统的强酸、强碱催化剂,也有别于过渡金属配合物的均相催化体系。为了更清楚地理解此类催化剂的催化机理,以促进此类催化剂的发展,本文将从过渡金属的种类、催化剂的制备方法等方面对此类催化剂进行总结。值得一提的是,近年来负载纳米单质金属(包括钯、金和银)催化剂在腈水合生成酰胺反应中也多见报道,最近已有全面综述[18],因此本文不再涉及。
1 过渡金属氧化物催化腈水合生成酰胺
1.1 二氧化锰催化剂
二氧化锰(MnO2)来源和应用广泛,常用作选择性氧化反应的氧化剂。研究表明 MnO2还可作为催化剂催化腈水合生成酰胺[19-24]。纯的MnO2粒度细小,因此很难与产物分离,为解决这一难题,NIE等[25]将其负载于氧化石墨烯上,制得负载型MnO2(MnO2/GO),MnO2以5~20 nm半径、100~600nm长度的棒状形态均匀、紧密地附着于氧化石墨烯的表面。与单纯的 MnO2相比,MnO2/GO是一种更有效的催化剂,可使包括脂肪醇、芳香醇、杂芳香醇以及烯丙醇等各种结构的醇与氨水反应生成酰胺,而在醇与氨反应转化为酰胺过程中,由醇生成的腈的水合是整个反应的关键基元步骤。
为了克服釜式反应中 MnO2与产物的分离难题,也有人采用固定床反应器进行腈的水解生成酰胺[26]。表1给出了不同结构的腈在固定床反应器上水合生成酰胺反应的结果。由表1中结果可知,不同结构腈的水合反应的活性顺序为:杂芳基腈>脂肪腈>芳基腈~乙烯基腈。对于杂芳基腈,反应物而言,杂原子与氰基越近,反应越容易进行,且杂环上吸电子基促进反应进行。就芳基腈而言,芳环上的吸电子取代基促进反应的进行,且吸电子能力越强,反应越快,因为吸电子基加强了氰基碳的电正性;给电子基不利于反应的进行,因为给电子基降低了氰基碳的电正性。同时,氰基邻位的取代基由于空间位阻不利于反应的进行。
腈水合反应除了与反应物结构有关外,ROY 等[21]还发现,采用不同方法制备的MnO2在3-氰基吡啶水合生成烟酰胺反应中具有不同的催化活性。表2给出了不同制备方法制备的MnO2在3-氰基吡啶水合生成烟酰胺反应中的催化性能。
由表2可知,由氯化锰与高锰酸钾在室温反应制备的MnO2具有最大比表面积和酸性,其催化活性最高;而采用硝酸锰高温分解得到的MnO2比表面积很低,尽管其酸性并不是很低,其催化活性最差。很显然,MnO2作为腈水合反应催化剂的关键是其应具有高的比表面积。腈水合生成酰胺依赖于MnO2的结构及形貌特征现象,还可以由基于八面体分子筛状的二氧化锰(OMS-2)可以顺利催化醇的氨化-水合合成酰胺反应,而其他MnO2的活性很低这一事实得到佐证[27-28]。
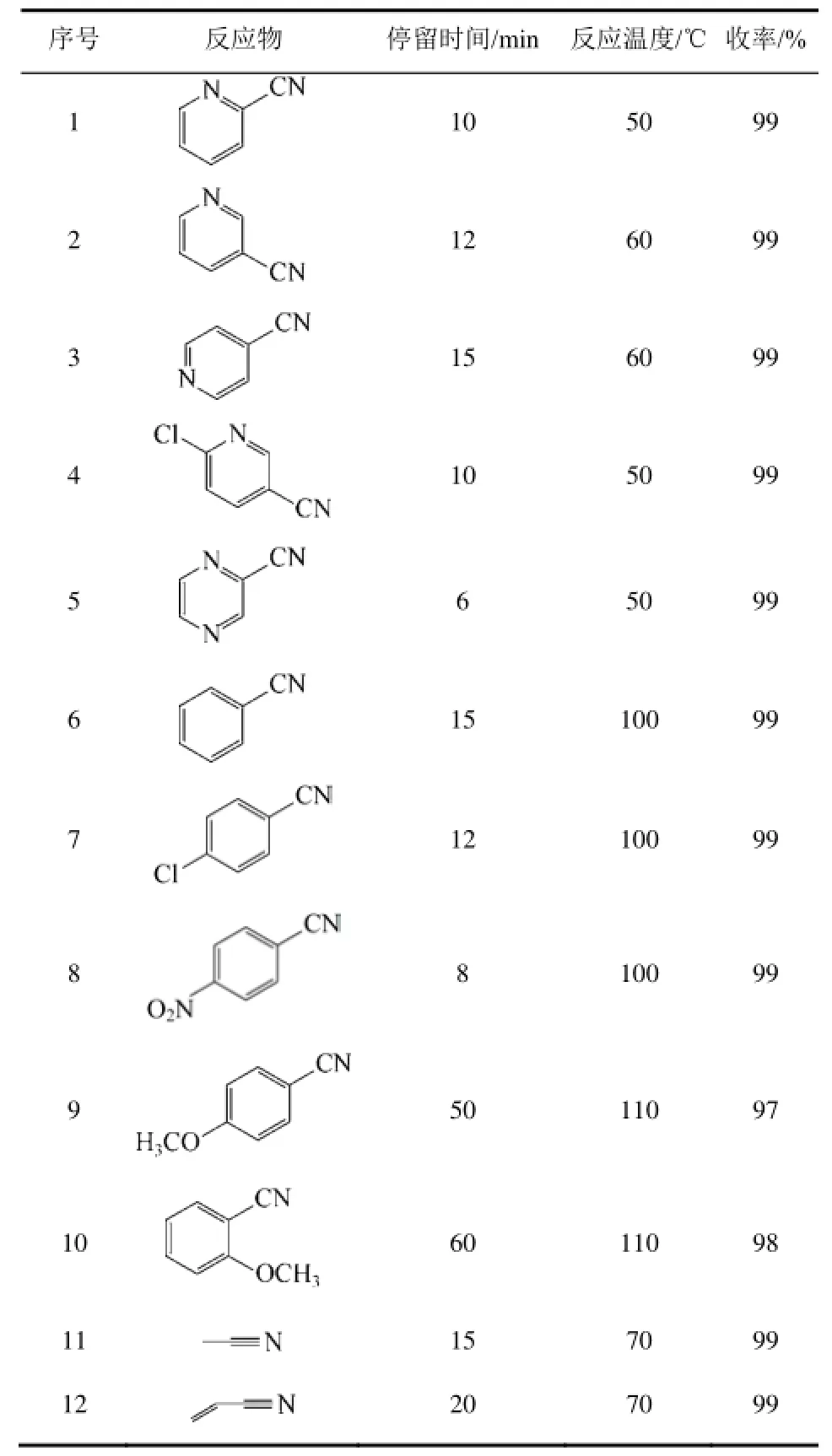
表1 固定床反应器中不同结构腈水合生成酰胺[26]
根据催化反应和 MnO2的表征结果,提出了MnO2催化腈水合生成酰胺反应机理[21,29]:MnO2的催化活性源自其在富水条件下形成表面羟基,这些表面羟基根据金属-氧键(Mn—O)的极化程度既可以显酸性也可以显碱性。极化的转变决定于所涉及的金属的电荷与半径、金属-氧键的键长以及金属离子的配位状态、金属d轨道的填充状态以及周围配体的性质。对于一个双配位的羟基金属离子而言,两个正金属离子(Mn4+)中心从氧上吸电子从而减弱氧-氢键(O—H),使得氢离子可以电离进入溶液,因而产生酸性羟基。酸性羟基被多个水分子包围。反应开始,首先腈分子吸附于酸性羟基上,进而氰基CN氮原子的孤对电子与MnO2的表面羟基通过静电吸引形成配位键,然后一个水分子的羟基(—OH)进攻质子化氰基,由水分子解离下的质子与 MnO2上的羟基负离子(—O—)结合再生酸性羟基。通过互变异构亚胺酸转化为酰胺,并从 MnO2表面脱附。由图 1可以看到,水分子对质子化的氰基的进攻进而发生水合作用是反应的关键步骤。很显然,腈分子中的吸电子基可使质子化氰基的电正性增强,促进反应的进行(表1的序号6~8);而给电子基则降低质子化氰基的电正性,不利于反应的进行(表1的序号9,10)。
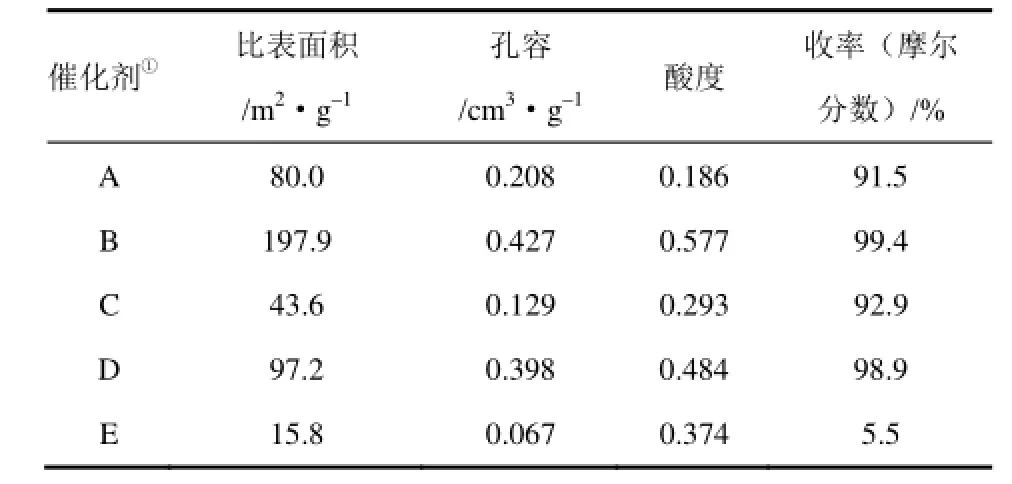
表2 不同方法制备的MnO2催化剂的性质与活性[21]
1.2 氧化镍催化剂
SAKAI等[30]最早将氧化镍(NiO)用作腈水合制备酰胺反应的催化剂。实验证明NiO只对杂芳基腈的水合有良好的催化活性,而对其他腈的水合则几乎没有催化活性。例如在NiO存在下,3-氰基吡啶和4-氰基吡啶在水溶液中回流反应7h,相应酰胺的收率可达90%。但是,当反应物为2-氰基吡啶时,反应物与镍形成配合物阻止反应的进行。后来,SUGIYAMA等[31]分别采用硝酸镍和碱式碳酸镍高温分解制备NiO,将其用于丙烯腈水合合成丙烯酰胺,但收率仍很低。自此,很长一段时间以氧化镍催化腈水合合成酰胺的研究未见诸文献。

图1 MnO2催化丙烯腈水合生成丙烯酰胺反应的机理[21,29]
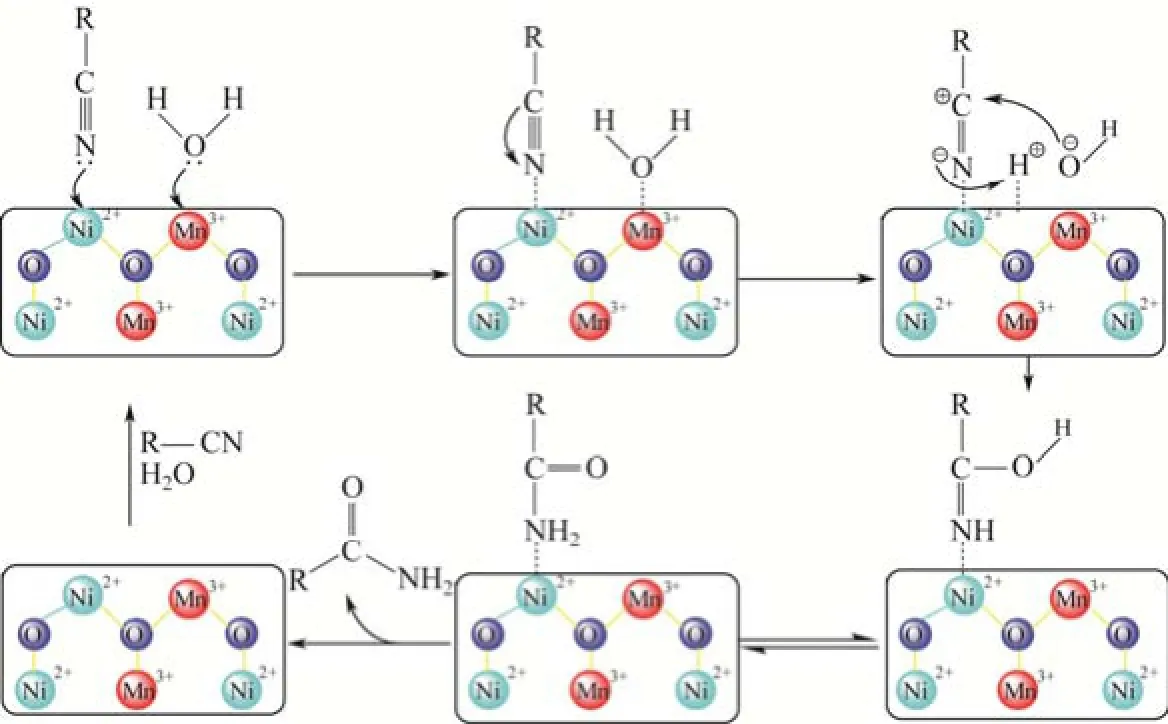
图2 Ni0.7Mn0.3O催化腈水合生成酰胺反应的机理[32]
最近,GOPAL等[32]通过向NiO中掺杂第二金属组分(Cu、Co和Mn等),获得了系列双金属组分氧化物Ni1-xMxO催化剂,将这些双金属氧化物用于苯甲腈的水合反应,显示出优良的催化性能。其中Ni0.7Mn0.3O 的催化活性最佳,当苯甲腈 5mmol、Ni0.7Mn0.3O 0.4g,在200mL水中于140℃反应15h,苯甲酰胺的收率高达99%。将Ni0.7Mn0.3O用于其他腈的水合,也取得了很好的结果(表3)。芳基上的取代基对反应的影响与MnO2为催化剂时结果相似。
GOPAL等[32]对 Ni0.7Mn0.3O的催化机理进行了探索。由于NiO自身对苯甲腈水合反应的催化活性很低,而Ni0.7Mn0.3O的催化活性较高,很显然 Mn在双金属氧化物催化中起重要作用。通过电子能谱分析发现处于Ni0.7Mn0.3O表面的Mn以Mn3+形式存在。由于 Mn3+具有比 Ni2+和 Mn2+强得多的酸性位,使得其成为活性中心,强化了反应介质中的水分子、氰基与催化剂的键合。因此,他们认为Ni0.7Mn0.3O催化的腈水合生成酰胺反应的机理包括如下步骤(图2):①首先水分子通过O原子与Mn3+结合,然后发生O—H断裂使水分子解离,金属氧化物晶格中相邻氧原子与氢原子间形成氢键促进了这种解离;②腈通过氰基配位子的亲核进攻。因此,双金属氧化物表面的金属离子对水分子和腈分子的共同活化,促进了腈水到 Ni2+;③OH–向吸附于相邻金属位点上氰基碳原合生成酰胺反应的进行。根据该机理并结合表3的反应结果,可知苯环上吸电子基有利于反应的进行,给电子基不利于反应的进行。另外,氰基邻位有取代基时,由于空间位阻,也不利于反应的进行。
1.3 氧化铈催化剂
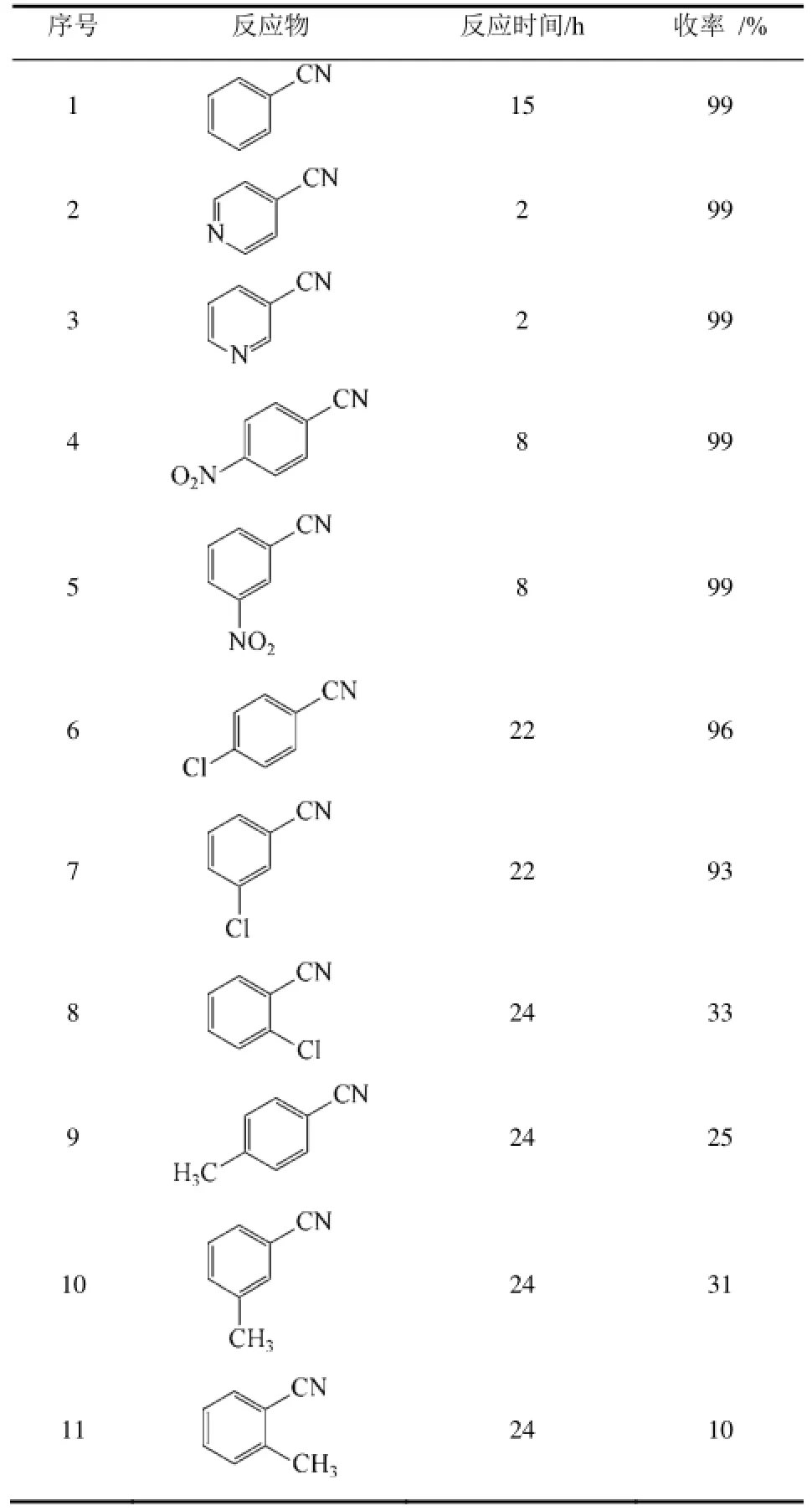
表3 Ni0.7Mn0.3O催化腈水合生成酰胺[32]
二氧化铈(CeO2)具有酸-碱性以及氧化-还原性,因此可用作多种反应的高效催化剂。这些反应包括醇的脱水、芳香化合物的烷基化、醇的二聚、二醇的环合、羧酸的还原以及由醛、酯、羧酸合成酮。除上述反应外,CeO2还用作腈水合、醇解以及胺解反应的催化剂[33]。SHIMIZU等[34]首次将CeO2用作腈水合生成酰胺反应的催化剂。初步研究发现,CeO2只对氰基邻位含杂原子(N 和O)腈的水合有催化活性,而对其他腈的水合几乎没有催化活性(表4)。
随后,SHIMIZU等[35]利用甲醇吸附红外分析方法确定了催化活性与低配位铈原子位点(CeLC,即氧缺陷位点)及相邻路易斯碱(暴露氧)的量之间的关系,揭示出 CeLC—O 位点是反应的活性位。

表4 不同条件下CeO2催化各种结构的腈水合生成酰胺[34]
他们还采用原位红外技术结合动力学同位素效应对反应机理进行了研究,提出了如图3所示的催化循环:①水分子H2O在CeLC—O位点(氧缺陷位点)上解离成OHδ–和Hδ+;②氰基邻位的杂原子如氮与Ce原子配位使腈分子与CeO2之间形成催吸附络合物;③OHδ–加成到络合物氰基的碳原子上;④酰胺分子从 CeO2表面脱附同时完成CeLC—O位点的再生。反应过程中氰基邻位杂原子与Ce原子的配位是反应得以进行的关键,因而不含杂原子的腈不能被 CeO2催化水合生成酰胺。

图3 CeO2催化腈水合生成酰胺反应的机理[35]
将 CeO2负载于聚苯乙烯或聚甲基丙烯酸甲酯上,得到的有机-无机杂化纳米粒子对2-氰基吡啶水合生成2-吡啶甲酰胺反应的催化活性更高,而且循环实用性比单纯的 CeO2更好[36]。尽管CeO2不能催化苯甲腈水合生成酰胺(表4的序号8),而且由二氧化碳与甲醇直接反应生成碳酸二甲酯的平衡常数很低,但将苯甲腈水合与CO2甲酯化反应偶合时,既可以得到苯甲酰胺又可以得到碳酸二甲酯[37]。说明在特定条件下,不含杂原子的腈也能水合生成酰胺,有待进一步研究,如式(2)、式(3)。
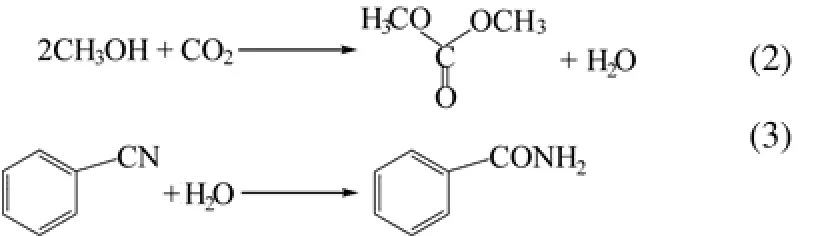
近来TAMURA等[38]又将CeO2拓展到腈、水以及伯胺的三组分“一锅”反应由腈直接合成N-烷基取代的酰胺,所试含杂原子腈几乎定量地转化为相应的N-烷基取代酰胺,如式(4)。

1.4 氧化钌催化剂
与前面的金属氧化物不同,氧化钌本身对腈的水合几乎没有催化活性。当将三氯化钌负载于三氧化二铝(Al2O3)载体,再经氢氧化钠处理后,得到负载钌催化剂[Ru(OH)x/Al2O3][39]。在该催化剂中,Ru(OH)x中的氢氧根数低于3,因而可认为该组分介于三氧化二钌与氢氧化钌之间。Ru(OH)x/Al2O3在许多反应中显示良好的催化活性[39-41]。MIZUNO等[42]将Ru(OH)x/Al2O3用于腈水合生成酰胺,在140℃的水溶液中可将各种腈高选择性地转换为相应的酰胺(表5)。
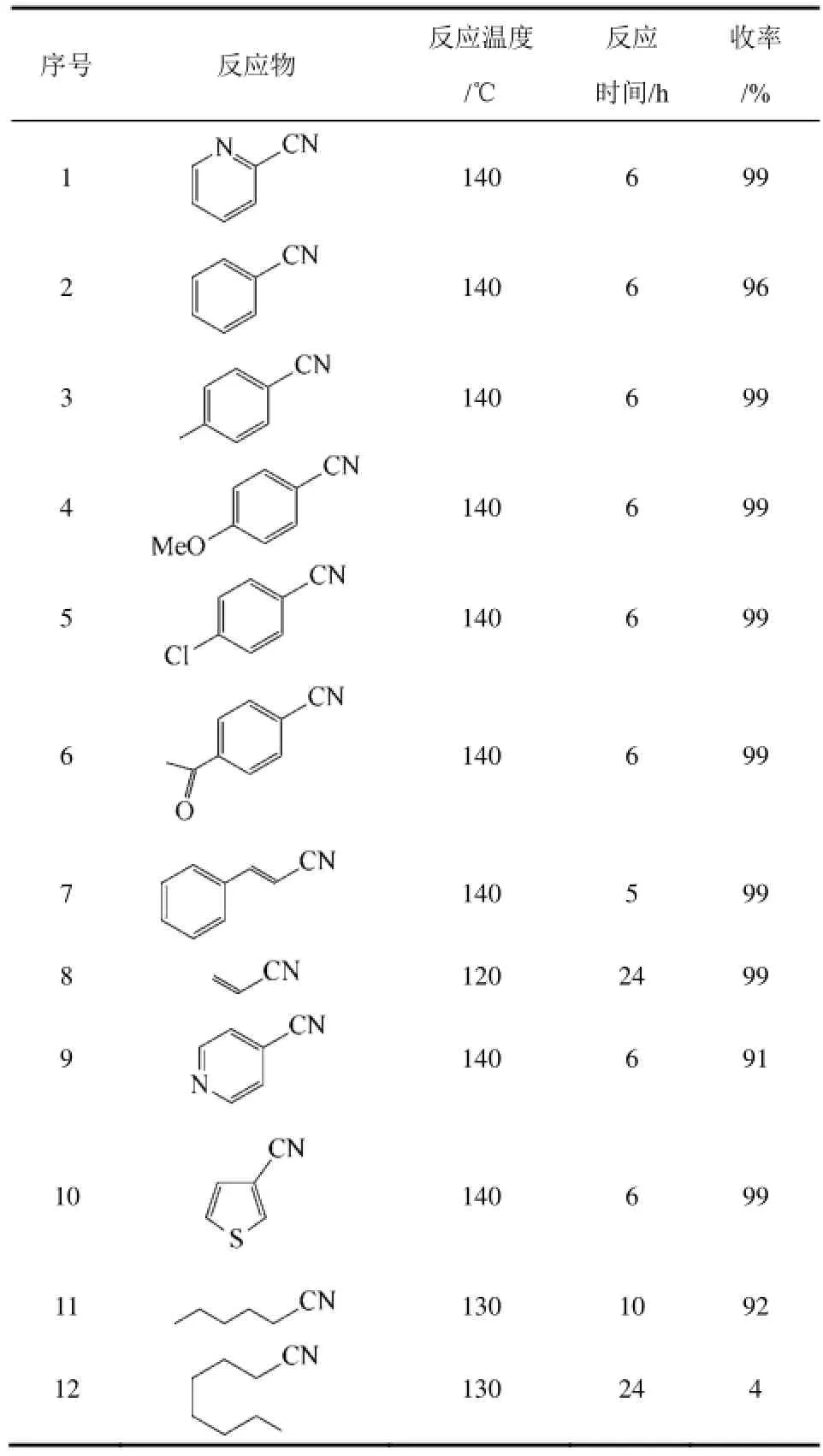
表5 Ru(OH)x/Al2O3催化不同结构腈水合生成酰胺[42]
将表5与表1、表3及表4相比,似乎可以看出,Ru(OH)x/Al2O3为催化剂时,含杂原子的腈并未比不含杂原子的腈水合的更快。对比苯甲腈与对位取代的苯甲腈的水合速率发现,p-OCH3(kX/kH=1.28)>p-CH3(1.01)>p-H(1.00)>p-Cl(0.79)>p-COCH3(0.70),其中,X为某甲腈对位上不同的取代基。这种Hammett值的对数与Brown-Okamoto值σ+之间的良好的线性关系表明Ru(OH)x/Al2O3催化的腈水合反应的机理与其他过渡金属氧化物催化的机理不同。根据这些结果作者提出了Ru(OH)x/Al2O3催化腈水合生成酰胺反应的可能机理(图4)。

图4 Ru(OH)x/Al2O3催化腈水合生成酰胺的反应机理[42]
Ru(OH)x/Al2O3催化的腈的水合反应由以下3步构成:首先腈分子以氰基中的氮原子与氢氧化钌(Ⅰ)中的钌中心配位形成中间体Ⅱ(第一步);然后,中间体II中的氢氧根物种亲核进攻氰基中的碳原子,生成钌亚胺盐中间体Ⅲ或钌与酰胺形成的η2-酰胺配合物Ⅳ(第二步),第二步可能包含碳正离子类型的碳正离子;水分子与中间体Ⅱ或Ⅳ之间进行配体交换生成酰胺并再生氢氧化钌物种。在Ru(OH)x催化腈水合生成酰胺的反应中,氰基直接与Ru配位得到活化,受底物的结构影响较小,因此,除长链脂肪腈外,各种结构的腈都可以在近乎相同工艺条件下水合生成酰胺。

图5 纳米Fe3O4-Ru(OH)x催化剂的合成[43]

图6 一锅法合成纳米Fe@SiO2Ru催化剂[43]
后来,VARMA等[43]将Ru(OH)x分别负载于氨基修饰和二氧化硅包覆的磁性纳米粒子上,制得可磁性回收的催化剂。这些催化剂在腈水合生成酰胺反应中均显示优良的催化性能。催化剂的制备过程分别如图5和图6所示。首先制备Fe3O4磁性纳米粒子,然后用多巴胺对其表面进行氨基修饰或用四乙氧基硅通过溶胶-凝胶方法在其表面包覆二氧化硅壳层,进而通过 RuCl3水解将Ru(OH)x负载其上。

图7 壳聚糖负载Ru(OH)x的合成[44]
最近,VARMA等[44]又将Ru(OH)x负载到壳聚糖分子上,制备了天然有机高分子负载的Ru(OH)x催化剂(图 7),该催化剂对各种腈水合生成酰胺具有优良的催化活性和选择性。该催化剂的优点是制备便捷,而且由于壳聚糖的亲水性,使催化剂活性更高。
1.5 其他过渡金属氧化物催化剂
其他各种过渡金属氧化物都曾被尝试用于腈水合生成酰胺反应的催化剂。Fe2O3、Co2O3、CuO 和ZnO曾被用作苯甲腈水和生成酰胺反应的催化剂。当反应在225℃反应时,Fe2O3催化苯甲腈水解生成酰胺,而CuO和ZnO由于与产物苯甲酰胺作用很快失活[32]。CuO、Fe2O3、WO3、Ag2O、ZnO、PbO、TiO2、La2O3和Co3O4在丙烯酰胺水和生成丙烯酰胺反应中也显示较低的活性[31];而TiO2、Y2O3、La2O3和ZrO2可催化2-氰基吡啶水合生成2-吡啶甲酰胺,但活性较低[34]。目前尚未见其他过渡金属氧化物有效催化腈水合生成酰胺反应的报道。
2 结 语
采用过渡金属氧化物催化腈水合生成酰胺具有原子经济性高、对环境友好的特点。由于过渡金属氧化物催化腈类水合的机理各不相同,并非每一种催化剂对所有腈的水合都有优良的催化性能,因此,应针对不同结构的腈选用不同的过渡金属氧化物催化剂。尽管此类催化剂较传统的酸碱催化剂具有对环境友好、反应的选择性高的优点,但也存在自身的不足,表现在:由于腈水合反应的过渡态能量较高,此类催化剂催化的腈水合反应仍然较高(一般在140℃甚至更高),因此,不适合分子结构共存其他更活性基团的腈的水合;有些催化剂由于粒度较小,与产物的分离困难;一些催化剂反应物的适用面太窄。基于此,未来此类催化剂应向复合型方向发展,即将不同的过渡金属氧化物前体按不同比例组合进而转化为多组分过渡金属氧化物,以拓宽反应物的适用范围。为解决分离问题,可以通过浸渍、共沉淀等方法将过渡金属氧化物活性组分负载于各种载体之上,得到负载型催化剂。由于这些催化剂不仅对腈水合反应有催化作用,还对其他反应如醇的脱氢、氧化有催化活性,因此可将醇的脱氢-氨化与水合反应串联,由更廉价易得的醇直接合成酰胺。
参考文献
[1] MABERMANN C E.Encyclopedia of Chemical Technology[M].KROSCHWITZ J I eds.New York:Wiley,1991:251-266.
[2] LIPP D.Encyclopedia of Chemical Technology[M].KROSCHWITZ.J I eds.New York:Wiley,1991:266-287.
[3] OPSAHL R. Encyclopedia of Chemical Technology[M].KROSCHWITZ J I eds.New York:Wiley,1991:346-356.
[4] LAROCK R C.Comprehensive organic transformations[M].2nd ed.New York:Wiley-VCH,1999.
[5] LOUDON M G.Organic Chemistry[M].New York:Oxford University Press,2002:982-983.
[6] CONSTABLE D J C,DUMN P J,HAYLER J D,et al.Key green chemistry research areas——a perspective from pharmaceutical manufacturers[J].Green Chem.,2007,9:411-420.
[7] 马文婵,周乔,张月成,等.醇-胺直接脱氢及氧化脱氢偶联酰胺化反应[J].化学进展,2014,26(2/3):334-344.
[8] GANGARAJULA Y,GOPAL B.Investigation of nano NiO, supported and metal ion substituted NiO for selective hydration of aromatic nitriles to amides[J].Appl.Catal.A:General,2014,475:211-217.
[9] GARCÍA-ÁLVAREZ R, CROCHET P, CADIEMNO V.Metal-catalyzed amide bond forming reactions in an environmentally friendly aqueous medium: nitrile hydrations and beyond[J].Green Chem.,2013,15:46-66.
[10] HU Y,JIN S,ZHANG Z,et al.One-step synthesis of nitriles by the dehydrogenation–amination of fatty primary alcohols over Cu/m-ZrO2[J].Catal.Commun.,2014,54(5):45-49.
[11] ZHANG YN,ZHANG YC,FENG C,et al.Amination of ethanol toacetonitrile over Ni-doped Co/γ-Al2O3catalyst[J].Catal.Commun.,2009,10(10):1454-1458.
[12] FENG C,ZHANG Y,ZHANG Y,et al.Study on alumina-supported cobalt-nickel oxide catalyst for synthesis of acetonitrile from ethanol[J].Catal.Lett.,2011,141(1):168-177.
[13] DAMODARA D,ARUNDHATHI R,LIKHAR P R.Copper nanoparticles from copper aluminum hydrotalcite:an efficient catalyst for acceptor- and oxidant-free dehydrogenation of amines andalcohols [J].Adv.Synth.Catal.,2014,356:189-198.
[14] KAWAGOE Y,MORIYAMA K,TOGO H.One-pot transformation of methylarenes into aromatic nitriles with inorganic metal-freereagents
[J].Eur.J.Org.Chem.,2014,2014(19):4115-4122.
[15] OISHI T,YAMAGUCHI K,MIZUNO N.Catalytic oxidative synthesis of nitriles directly from primary alcohols and ammonia[J].Angew.Chem.Int.Ed.,2009,48:6286-6288.
[16] RYLAND B L,Stahl S S.Practical aerobic oxidations of alcohols and amines with homogeneous copper/TEMPO and related catalystsystems [J].Angew.Chem.Int.Ed.,2014,53:8824-8838.
[17] TAKIYA J A,KNAPP S M M,TYLER D R.Frontiers in catalytic nitrile hydration:nitrile and cyanohydrin hydration catalyzed byhomoge neous organometallic complexes[J].Coord.Chem.Rev.,2011,255:949-974.
[18] DOWNS E L,TYLER D R.Nanoparticle catalysts for nitrile hydration[J].Coord.Chem.Rev.,2014,280:28-37.
[19] HAEFELE L R,YOUNG H J.Catalyzed hydration of nitriles to amides[J].Ind.Eng.Chem.Prod.Res.Dev.,1972,11:364-365.
[20] ROY S K,RAY S C,RAY P K.The catalyzed hydration of nitriles to amides[J].J.Indian Chem.Soc.,1980,57:195-198.
[21] ROY S C,DUTTA P,NANDY L N,et al.Hydration of 3-cyanopyridine to nicotinamide over MnO2catalyst[J].Appl.Catal.A:General,2005,290:175-180.
[22] SHEN C H,LEE C Y,TSAI C J.Process for producing organic carboxylic acid amides PCT Int.Appl.:US20110004020[P].2011-01-06.
[23] YAMAGUCHI K,WANG Y,KOBAYASHI H,et al.Efficient hydration of nitriles promoted by simple amorphous manganese oxide using reduced amounts of water[J].Chem.Lett.,2012,41:574-576.
[24] YAMAGUCHI K,WANG Y,MIZUNO N.Manganese oxide-catalyzed additive- and solvent-free aerobic oxidative synthesis of primary amides from primary amines[J].Chem.Lett.,2012,41:633-635.
[25] NIE R F,SHI J J,XIA S X,et al.MnO2/graphene oxide:a highly active catalyst for amide synthesis from alcohols andammonia in aqueous media[J].J.Mater.Chem.,2012,22:18115.
[26] BATTILOCCHIO C,HAWKINS J M,LEY S V.Mild and selective heterogeneous catalytic hydration of nitriles to amides by flowing through manganese dioxide[J].Org.Lett.,2014,16(4):1060-1063.
[27] YAMAGUCHI K,KOBAYASHI H,OISHI T,et al.Heterogeneously catalyzed synthesis of primary amides directly from primary alcohols and aqueous ammonia[J].Angew.Chem.,2012,124:559-562.
[28] YAMAGUCHI K,KOBAYASHI H,WANG Y,et al.Green oxidative synthesis of primary amides from primary alcohols or aldehydescatalyzed by a cryptomelane-type manganese oxide-based octahedral molecular sieve,OMS-2[J].Catal.Sci.Technol.,2013,3:318-327.
[29] SUGIYAMA K,MIURA H,NAKANO Y,et al.Liquid-phase hydration of acrylonitrile to acrylamide over the manganese dioxide catalyst[J].Bull.Chem.Soc.Jpn.,1987,60:453-456.
[30] SAKAI K,ITO T,WATANABE K.Studies of organic catalytic reactions.V.The function of the chelates formed in the hydrolysis of 2-cyanopyridine with metal oxide catalysts[J].Bull.Chem.Soc.Jpn.,1967,40:1660-1665.
[31] SUGIYAMA K,MIURA H,NAKANO Y,et al.Heterogeneous hydration of acrylonitrile over the metal oxide catalysts in liquid phase[J].Bull.Chem.Soc.Jpn.,1986,59:2983-2989.
[32] GANGARAJULA Y,GOPAL B.Investigation of nano NiO, supported and metal ion substituted NiO for selective hydration of aromatic nitriles to amides[J].Appl.Catal.A:General,2014,475:211-217.
[33] TAMURA M,SHIMIZU K,SATSUMA A.CeO2-catalyzed transformations of nitriles and amides[J].Chem.Lett.,2012,41(11):1397-1405.
[34] TAMURA M,WAKASUGI H,SHIMIZU K,et al.Efficient and substrate-specific hydration of nitriles to amides in water by using a CeO2catalyst[J].Chem.Eur.J.,2011,17:11428-11431.
[35] TAMURA M,SATSUMABC A,SHIMIZU K.CeO2-catalyzed nitrile hydration to amide:reaction mechanism and active sites[J].Catal.Sci.Technol.,2013,3:1386-1393.
[36] MARI M,MULLER B,LANDFESTER K,et al.Ceria/polymer hybrid nanoparticles as efficient catalysts for the hydration of nitriles to amides[J].IACS Appl.Mater.Interfaces,2015,7(20):10727-10733.
[37] HONDA M,KUNO S,SONEHARA S,et al.Tandem carboxylation-hydration reaction system from methanol,CO2and benzonitrile to dimethyl carbonate and benzamide catalyzed by CeO2[J].Chem.Cat.Chem.,2011,3:365-370.
[38] TAMURA M,TONOMURA T,SHIMIZU K,et al.CeO2-catalyzed one-pot selective synthesis of N-alkyl amides from nitriles,amines and water[J].Appl.Catal.A:General,2012,417/418:6-12.
[39] YAMAGUCHI K,MIZUNO N.Supported ruthenium catalyst for the heterogeneous oxidation of alcohols with molecular oxygen[J].Angew.Chem.Int.Ed.,2002,41(23):4538-4542.
[40] YAMAGUCHI K,MIZUNO N.Efficient heterogeneous aerobic oxidation of amines by a supported ruthenium catalyst[J].Angew.Chem.Int.Ed.,2003,42(13):1480-1483.
[41] YAMAGUCHI K,MIZUNO N.Scope,kinetics,and mechanistic aspects of aerobic oxidations catalyzed by ruthenium supported on alumina[J].Chem.Eur.J.,2003,9:4353-4361.
[42] YAMAGUCHI K,MATSUSHITA M,MIZUNO N.Efficient hydration of nitriles to amides in water,catalyzed by ruthenium hydroxide supported on alumina[J].Angew.Chem.,2004,116(12):1602-1606.
[43] POLSHETTIWAR V,VARMA R S.Nanoparticle-supported and magnetically recoverable ruthenium hydroxide catalyst:efficient hydration of nitriles to amides in aqueous medium[J].Chem.Eur.J.,2009,15:1582-1586.
[44] NASIR BAIG R B,NADAGOUDA M N,VARMA R S.Ruthenium on chitosan:a recyclable heterogeneous catalyst for aqueous hydration of nitriles to amides[J].Green Chem.,2014,16(4):2122-2127.
第一作者:赵晓甫(1988—),男,硕士研究生。联系人:赵继全,教授。E-mail zhaojq@hebut.edu.cn。
中图分类号:TQ 032.4
文献标志码:A
文章编号:1000-6613(2016)07-2071-10
DOI:10.16085/j.issn.1000-6613.2016.07.019
收稿日期:2015-10-19;修改稿日期:2015-12-16。
基金项目:国家自然科学基金(21476057)及河北省自然科学基金(B2015202284)项目。
Progress on hydration of nitriles to amides catalyzed by transition metal oxides
ZHAO Xiaofu,ZHANG Yuecheng,ZHANG Hongyu,ZHAO Jiquan
(School of Chemical Engineering and Technology,Hebei University of Technology,Tianjin 300130,China)
Abstract:Hydration of nitriles to the corresponding amides has the advantages of high atomic economy and avoiding formation of undesirable side products.Traditionally,the hydration of nitriles was achieved by the catalysis of strong acid or alkali.However,it has the drawbacks of over hydrolysis of amides into carboxylic acids and the formation of salts from the neutralization of the catalysts.To overcome these difficulties various transition metal oxides including manganese dioxide (MnO2),nickel oxide (NiO),ceria (CeO2) and ruthenium hydroxide [Ru(OH)x] to replace strong acid or alkali have been used as catalysts in this reaction.In this article,the hydration of various nitriles to the corresponding amides catalyzed by the above transition metal oxides has been summarized.It can be seen that the hydration of nitriles to the amides depends on the type and preparation method of catalysts and the structure of nitriles.The preparation method,application scope,advantages and disadvantages of each catalyst are also illustrated.In addition the possible catalytic mechanisms of the catalysts are discussed.It is expected that the catalysts with excellent performance will be from complex transition metal oxides and supported metal oxides based on the above discussions.
Key words:hydration;nitriles;amides;transition metal oxides;manganese dioxide;nickel oxide;ceria;ruthenium hydroxide
猜你喜欢
河南化工(2021年4期)2021-05-12
昆明医科大学学报(2021年2期)2021-03-29
腐植酸(2020年5期)2020-12-20
四川警察学院学报(2019年6期)2019-12-28
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
World Journal of Clinical Cases(2019年4期)2019-04-16
文化产业(2016年6期)2016-10-19
火炸药学报(2014年3期)2014-03-20
食品工业科技(2014年13期)2014-03-11
