
Pim-1蛋白激酶与3,5-二取代吡唑并吡啶衍生物的分子识别研究
2016-07-22 06:24胡建平杜文义苟小军
成都大学学报(自然科学版) 2016年2期
梁 立, 胡建平,2, 杜文义, 左 柯, 刘 嵬, 苟小军
(1.成都大学 药食同源植物资源开发重点实验室, 四川 成都 610106;2.乐山师范学院 化学学院, 四川 乐山 614004)
Pim-1蛋白激酶与3,5-二取代吡唑并吡啶衍生物的分子识别研究
梁立1, 胡建平1,2, 杜文义1, 左柯1, 刘嵬1, 苟小军1
(1.成都大学 药食同源植物资源开发重点实验室, 四川 成都610106;2.乐山师范学院 化学学院, 四川 乐山614004)
摘要:具有丝/苏氨酸激酶活性的Pim-1蛋白激酶(Pim-1 protein kinase,PPK)的过度表达能导致肿瘤的发生,是一个有效的抗肿瘤药物作用靶点.对PPK与3,5-二取代吡唑并吡啶衍生物(3,5-Disubstituted pyrazolopyridine derivatives,DPD)的复合物进行了20 ns的分子动力学模拟,从能量角度分析了二者结合的主要驱动力以及I 185在识别过程中的重要作用,并对复合物体系在模拟过程中的构象变化进行了分析.结果表明,PPK的核心区的保守性是其发挥生理功能的重要机制,模拟结果对基于PPK抑制剂的药物设计具有一定参考作用.关键词: Pim-1蛋白激酶;分子动力学模拟;成簇分析;自由能曲面图;构象变化
0引言
癌症亦称恶性肿瘤,是一类由控制细胞生长增殖机制失常而引起的疾病,是目前威胁人类健康的主要疾病之一[1].研究发现,蛋白激酶通过磷酸化调控蛋白质活性,在细胞信号转导过程中发挥重要作用,其活性异常或过度表达会引发癌症、糖尿病、炎症神经退行性疾病等[2-3].因此,在使用小分子化合物对肿瘤治疗的研究方面,蛋白激酶已成为一个重要的靶点[4].Pim基因最早是作为莫洛尼小鼠白血病病毒的前病毒而被人们发现[5].Pim家族包括Pim-1、Pim-2和Pim-3,它们均具有丝/苏氨酸激酶活性的原癌基因,都包含一个被称为ATP锚的活化位点,属于钙/钙调蛋白调节激酶,在多细胞的进化过程中高度保守[6-7].结构生物学实验表明[5],PPK的催化结构域覆盖了从Y 38~M 290的氨基酸区域,这个区域中G 45~G 50段loop区是一个保守的甘氨酸环状模序,L 44~V 52以及K 67是磷酸盐结合位点,而D 167是一个质子受体位点.Bachmann等[8]发现,K67若突变为甲硫氨酸,则可导致PPK的失活性突变.PPK的作用机制是通过磷酸化不同的底物而促进细胞的生存和增殖,抑制细胞的凋亡,进而导致肿瘤的发生.
目前,分子动力学(Molecular dynamics,MD)可以在计算机上计算出分子的轨迹数据、结合模式以及能量和氢键等,用以推测和评价药物小分子与受体大分子相互作用的结合模式[9].Nishiguchi等[10]用X-ray方法解析出了PPK-DPD复合物的晶体结构,并证明DPD对PPK具有较好的抑制效果,但是他们之间的识别驱动力以及复合物体系在水溶液中的含水构象变化等问题尚不得知.对此,本研究采用MD模拟方法对上述问题展开研究,进而为研究DPD与PPK的识别机理提供帮助.图1为本研究的DPD衍生物的分子结构.
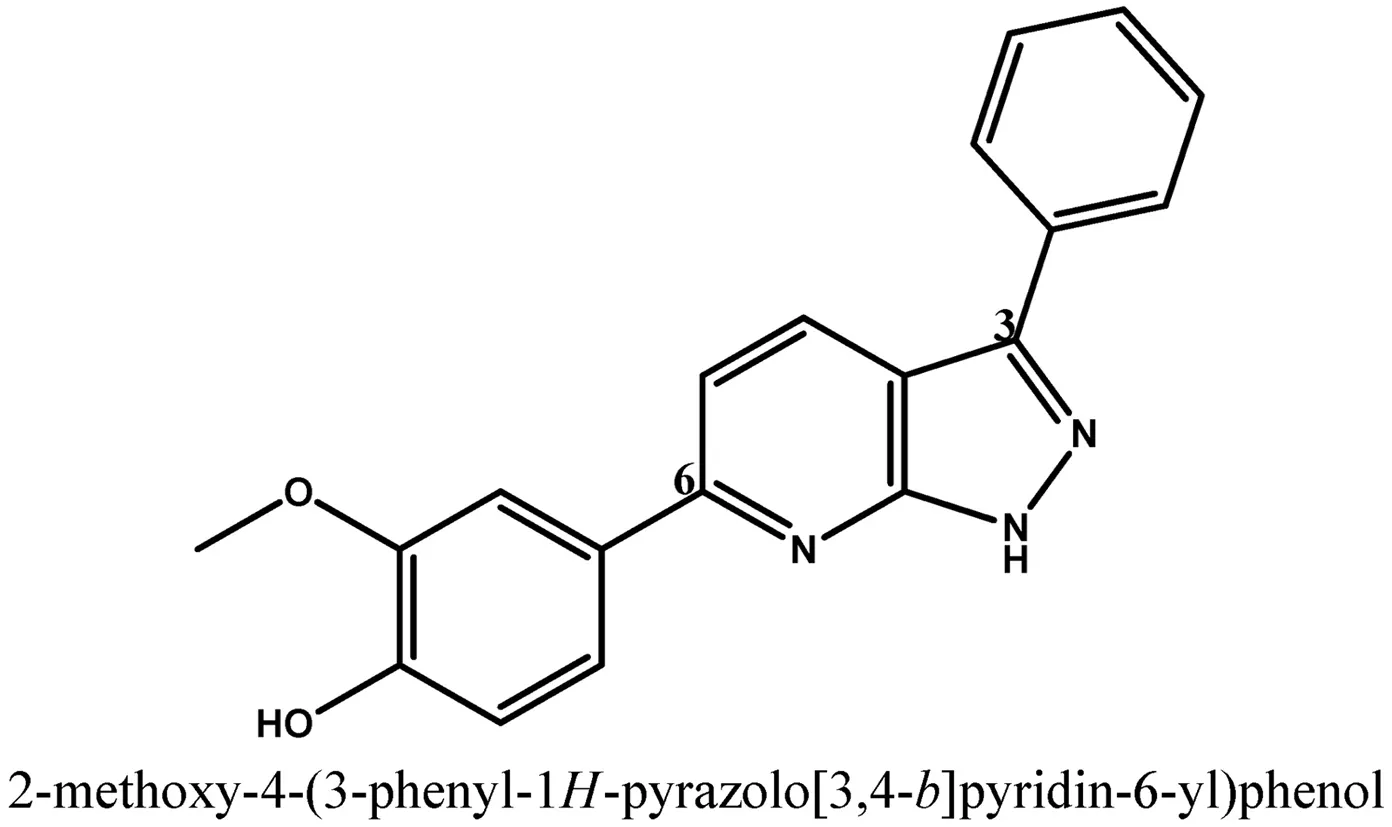
图1二取代吡唑并吡啶衍生物的分子结构
1计算方法
1.1MD模拟
基于PPK-DPD的复合物晶体结构(PDB代码:3T9I)[10],删除结晶水部分,仅保留A链和小分子DPD,以此作为MD模拟的初始构象.由于A链中的SEP261是一个含有磷酸根的非标准氨基酸(SEP),这将导致程序无法识别,但是通过观察PPK蛋白的空间结构,发现SEP 261~S 306的结构处于整个蛋白的外侧,并且远离蛋白中DPD结合的位置,对二者的识别基本无影响,可以将SEP 261~S 306的氨基酸删除,因此,用于MD模拟的蛋白质残基范围是P 33~S 260,共228个氨基酸.
MD模拟采用Amber程序[11],力场为Amber力场,蛋白质的力场参数均基于实验值拟合[12].模拟温度为300 K,溶剂采用TIP3P水模型[13].首先,在溶质抑制剂外围加上1.0 nm的去头八面体水盒子,总共加入7 921个水分子,含水后体系的总原子数为27 476个.在MD模拟之前,对体系进行了2次能量优化:首先,约束溶质(约束力常数为2.09×105 kJ/mol·nm2),用最陡下降法优化200步,再用共轭梯度法优化1 800步;其次,在去除所有约束后进行200步最陡下降法优化和1 800步共轭梯度法优化,收敛条件为能量梯度小于4.18×10-4kJ/mol·nm.MD.模拟分为2步:先进行2 ns约束溶质的MD模拟(约束力常数为4.18×103 kJ/mol·nm2),期间温度从0 K逐步升高到300 K;接着再进行18 ns的无约束恒温MD模拟.在模拟过程中,用VMD图像显示软件实时跟踪体系的构象,采用SHAKE算法[14]约束键长,MD模拟的积分步长设为2 fs,非键相互作用截断半径设为1 nm,每隔1 ps采集一次构象,共采集20 000个构象.
1.2能量分解
能量分解采用MM/GBSA(Molecular mechanics/generalized born surface area)方法[15],其基本思路是把每个残基的能量贡献近似分为用分子力学方法计算的真空分子内能,极性溶剂化能用广义波恩(GB)模型[16]计算,非极性溶剂化能用LCPO模型[17]计算,并且将能量分解到残基的主链原子和侧链原子上.据此,可以进一步考察蛋白质中的主要残基对结合物分子所产生的贡献,发现与结合物分子结合的主要位点.
1.3成簇分析
对体系MD模拟所获得的20 000个构象进行成簇分析,分析采用MMTSB工具[18],计算过程使用Daura等[19]的策略,其基本思路是:依次逐个计算20 000个构象的RMSD值,设定一个RMSD域值,将20 000个构象中RMSD处于该域值的构象归为一簇,并将每簇中的能量最低构象作为该簇的代表性构象.本研究对复合物的20 000个构象进行了RMSD域值为0.23 nm的成簇分析.
1.4自由能曲面
自由能曲面(Free energy landscape,FEL)表示在生物学过程中发生的动力学变化,它可以通过模拟过程中的体系最稳定状态(即自由能曲面图的最小值)和体系变化过程中的瞬时状态(即分界点)来分析[20].主成分分析(Principle component analysis,PCA)可以表示体系中最重要的动力学过程,并可用于绘制自由能曲面图[21],其定义为,
ΔG(x)=-kBTlnP(X)
式中,X为本征向量(PC),kB为Boltzmann常数,T为绝对温度,P(X)则为体系对PC的贡献值.在本研究中,PC1和PC2的计算是FEL分析的基础,并可以联合成簇分析来讨论体系的构象变化.
2结果与讨论
2.1分子整体的模拟分析
2.1.1体系势能.
PPK与DPD复合物体系势能随时间的变化曲线如图2所示.
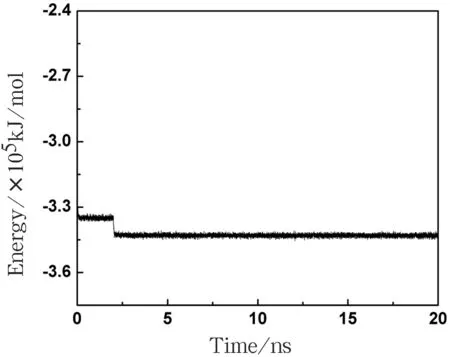
图2体系势能随时间变化
从图2可知,体系势能在2 ns后趋于平衡,在2 ns之前能量的平均值为-3.35×105kJ/mol,涨落幅度为1%,较大的涨落幅度与在模拟初始阶段溶剂化效应使得体系需要快速消除内部晶体堆积作用有关.体系能量在2 ns之后保持稳定,平均值为-3.43×105kJ/mol,波动范围为0.1%,对于具有27 476个原子的大体系来说,涨落误差可以忽略不计.由此可见,体系的整个模拟过程是平稳可靠的.
2.1.2方均根偏差.
PPK与DPD复合物的Cα原子方均根偏差(Root mean square deviation,RMSD)随时间的变化曲线如图3所示.

图3体系方均根偏差随时间的变化
从图3可知,在2 ns之前体系的Cα原子RMSD值稳定在较低的值,这是由于在MD模拟时首先对体系进行了2 ns的约束MD模拟,因此体系的运动幅度很小.在2~11 ns范围内,体系的RMSD值为0.260±0.061 nm,波动幅度为23%,体系的RMSD值迅速上升,这是由于体系从约束到无约束的过程中,体系原子会出现较大的振动幅度,在11 ns之后体系Cα原子的RMSD为0.293±0.014 nm,波动幅度约为4%,处于一个比较稳定的状态.另外,从图3中可以看出,体系的RMSD在11 ns前后出现了较大的波动,预示体系在11 ns前后可能有较为明显的构象变化过程.
2.1.3方均根涨落.
复合物体系整体Cα原子的方均根涨落(Root mean square fluctuation,RMSF)分布如图4所示.
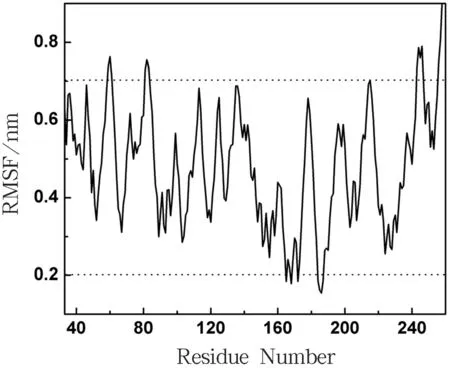
图4体系Cα原子的方均根涨落分布
从图4可知,涨落较大的区域为,S 59~N 61、P 81~G 83、Y 215、E 243~E 246和R 256~S 260,用VMD观察其在水溶液中的运动情况可以发现,S 59~N 61和P 81~G 83 2个区域分别处于2个β折叠之间的loop区,并且处于整个体系的外侧,充分暴露在水环境中;Y 215位于2个α螺旋之间,与周围的残基作用较弱;E 243~E 246是一段偏离质心较多的α螺旋,并且与R 256~S 260一样,处于整个链的末端,所以振动的幅度会相对较大.孙占莉等[22]认为,L 44~G 50的loop区是磷酸化的结合位点,据此可以推测S 59~N 61较大的运动幅度与PPK的磷酸化作用有关.另外,涨落较小的是H 165,I 168,N 172和L 184~F 187,观察发现,这些残基均处于PPK的中心位置,与周围残基的作用较大,因此运动幅度很小.尤其是L 184~F 187与DPD距离很近,推测其较低的振动幅度与DPD的结合有关.
RMSF和B因子均可用于反映体系在整个模拟过程中相对于平均构象所发生的变化,RMSF和B因子越大则对应残基的柔性越大.3T9I晶体结构B因子与MD模拟结果RMSF数值的相关性分析结果如图5所示.

图5晶体结构B因子与RMSF的相关性
从图5中可知,计算得到的RMSF与B因子的实验值的相关系数为0.35,由于样本量较大(N=228),因此2组数据存在显著的相关性,也证明整体柔性分析的可靠性.
2.2分子识别
2.2.1能量分解.
PPK与DPD的识别密切相关的残基的能量分解结果如表1所示.
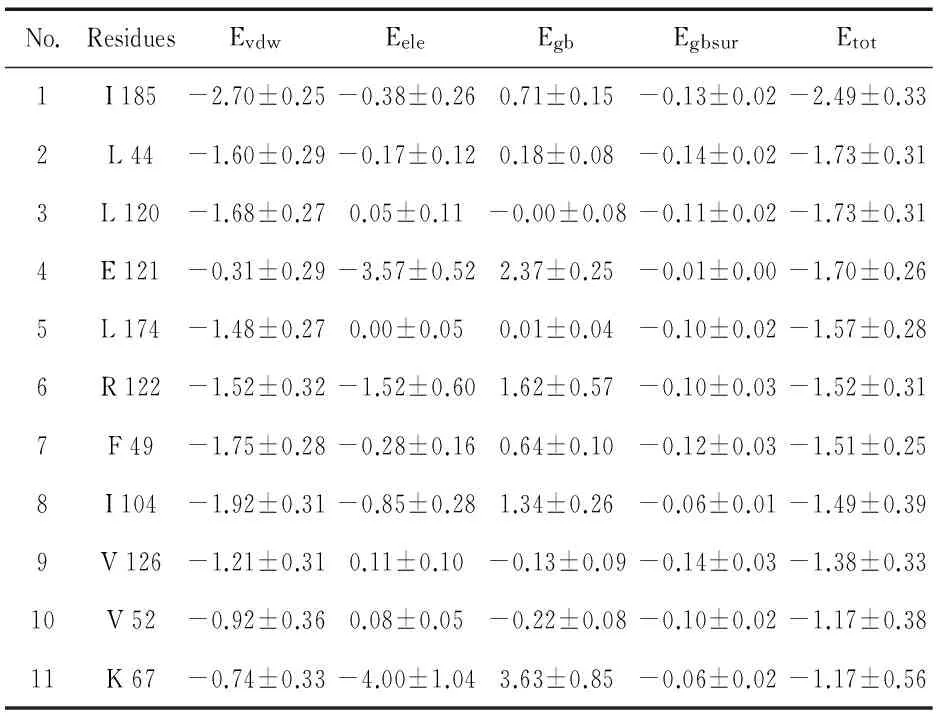
表1 PPK与DPD结合密切相关残基的能量分解结果(KJ/mol)
表1中,Evdw为真空状态下的范德华结合能,Eele为真空静电结合能,Egb为极性溶剂化结合能,Egbsur为非极性溶剂化结合能.从表1可知:分子内能(即Evdw+Eele)是促进二者结合的主要动力;PPK的G 45~V 52和L 120~V 126是有利于与抑制剂结合的主要区域.另外,I 185的能量最低,用VMD观察可知,I 185与DPD的空间位置非常接近,因此可以推测I 185在PPK与DPD的识别过程中起重要的作用,这与前面柔性分析的结果一致.
2.2.2结合模式.
DPD的接触残基情况如图6所示.接触残基定义为在4?范围内所有的氨基酸,DPD用紫色stick表示,其余残基均用绿色stick表示.

图6PPK与DPD的结合模式
从图6可以看出,DPD的接触残基共有17个,完全包含了上面能量分解结果中的所有残基,证明了上述能量分解的可靠性.并且上述的17个残基组成了一个“凹"形的口袋,DPD正好能够结合在该口袋中,也证明了这种结合模式的合理性.另外,从图6中可以看出,距离DPD最近的氨基酸有L 120,L 44,F 49,I 185和L 174,均为疏水性氨基酸,因此PPK与DPD识别的主要作用力为疏水作用.
2.2.3氢键.
PPK与DPD之间形成的氢键情况如表2所示.氢键采用几何判据[23],供体与受体之间的夹角(Angle)大于135°,供体与受体之间的距离(Distance)小于0.35 nm.表2中Freq表示氢键占有率,即指氢键在整个模拟过程中出现的几率.氢键从能量和方向性两个角度均有利于增强蛋白质与底物所形成复合物的稳定性,有利于二者间的识别.
从表2可以看出,R 57与D 60之间可形成了多个较稳定的氢键,通过观察二者在空间中的位置得知,R 57和D 60处于两个β折叠之间的loop区上,并且处于蛋白的最外侧,因此生理学意义不大.另外,Q 127与D 131位于整个结合口袋的外侧,推测二者之间的氢键对于维持整个口袋的入口形状起关键作用.
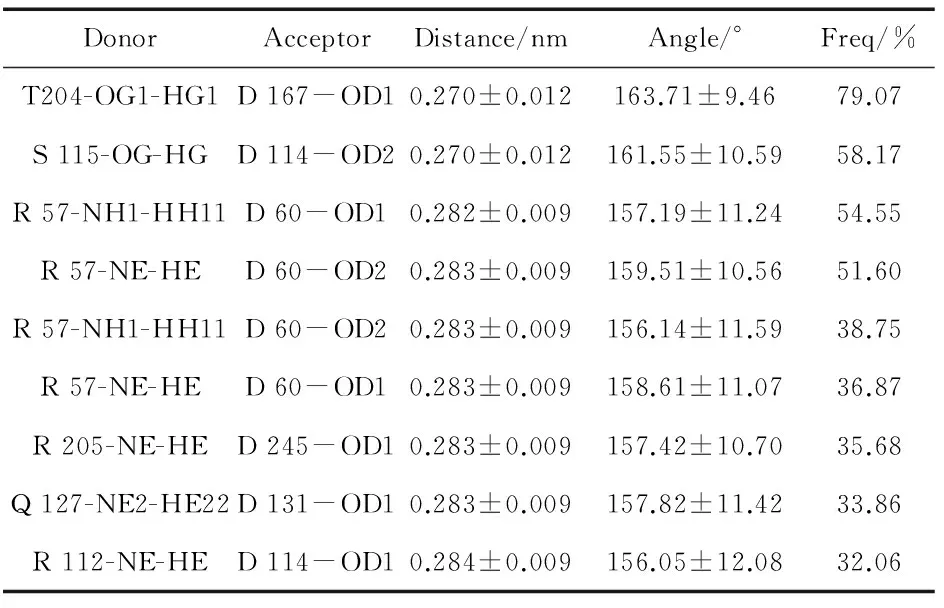
表2 PPK-DPD复合物内部的氢键
2.2.4关键残基的距离.
DPD与I 185的质心之间的距离随模拟时间的变化图如图7所示.

图7DPD与I 185之间的距离随时间的变化
从图7可以看出,二者之间的距离在11 ns后稳定,为0.574±0.025 nm,涨落幅度为4%,DPD与I 185处于较稳定的距离.表1中数据也显示I 185在DPD与PPK识别的过程中起到重要的作用,因此推测DPD-I 185之间的范德华力是PPK与DPD识别的关键.
2.3分子的运动性及构象分析
2.3.1自由能曲面.
PPK与DPD复合物体系的自由能面图如图8所示,图中颜色越深的区域,表示自由能越低.
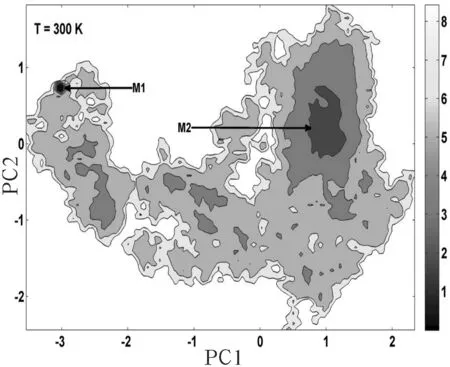
图8复合物的自由能曲面图
从图8可知,复合物体系有2个相对独立的能量区域,分别标记为M1和M2,这些独立的能量区域代表模拟过程中体系最稳定的状态.
2.3.2成簇分析.
为了更好研究图8中体系的能量最低构象,实验以2.3 nm为RMSD域值,体系的成簇分析结果如图9所示.

图9基于复合物体系的RMSD成簇分析
从图9可以看出,体系成簇数为2个,5.5 ns以前为第1簇,(5.5~20) ns为第2簇,它们所占的概率分别是28.0%和72.0%.显然,体系的构象在5.5 ns前后有较明显的变化,这与图3中RMSD的涨落情况相符合.
2.3.3构象分析.
为了更详细了解每簇的代表性构象,通过分析图2的势能结果,分别找到了2簇中的能量最低构象.图10给出了2个代表性构象的叠落图,对应的代码分别是4 736(绿色)和16 089(黄色).具体来说,体系构象变化不大, 小分子DPD的构象也没有发生较大变化,通过观察体系的整体构象得知,变化较大的是末端的I 249~S 260段,该段loop处于蛋白外侧,运动幅度较大,因此造成了RMSD波动较大,而整个体系的核心区域(即结合口袋附近,I 185,D 186和L 120~R 122等残基)的构象很稳定,未发生较大变化.因此,PPK核心区域较高的保守性是其发挥功能的关键.

图10 能量最低构象的叠落
3结论
通过对PPK-DPD复合物进行了20 ns的显含水MD分子动力学模拟,基于RMSF计算值与B因子实验值具有显著相关性,证明了MD模拟和柔性分析的可靠性.通过结合自由能和氢键的分析,给出了水溶液中PPK与DPD的结合模式图,得到DPD结合的主要驱动力是氨基酸的疏水作用的结论,并且提出I 185与DPD之间的范德华作用力在识别过程中起重要作用.通过自由能曲面和成簇分析体系构象变化发现,蛋白的核心区域并未发生较大的构象变化,因此,核心区域的保守性是PPK发挥功能的重要机制.本研究的模拟结果为更深入理解PPK与DPD的分子识别以及DPD类抑制剂的药物设计具有一定的指导意义.
参考文献:
[1]Ferlay J,Shin H R,Bray F,et al.Estimatesofworldwideburdenofcancerin2008:GLOBOCAN2008[J].International Journal of Cancer,2010,127(12):2893-2917.
[2]沈云婕,朱王君.新型分子靶向抗癌药物的研究进展[J].世界临床药物,2007,28(5):288-292.
[3]夏冬辉.原癌蛋白PIM-1激酶抑制剂的分子模拟研究[D].西安:西北大学,2012.
[4]Doudou S,Sharma R,Henchman R H,et al.InhibitorsofPIM-1kinase:acomputationalanalysisofthebindingfreeenergiesofarangeofimidazo[1,2-b]pyridazines[J].Journal of Chemical Information and Modeling,2010,50(3):368-379.
[5]Mochizuki T,Kitanaka C,Noguchi K,et al.Pim-1kinasestimulatesc-Myc-mediateddeathsignalingupstreamofcaspase-3 (CPP32)-likeproteaseactivation[J].Oncogene,1997,15(12):1471-1480.
[6]邓欢,刘亮明,张吉翔.丝/苏氨酸激酶Pim家族在细胞周期调控和肿瘤发生中的作用[J].国际遗传学杂志,2006,29(5):375-378+388.
[7]Mikkers H,Nawijn M,Allen J,et al.MicedeficientforallPIMkinasesdisplayreducedbodysizeandimpairedresponsestohematopoieticgrowthfactors[J].Molecular and Cellular Biology,2004,24(13):6104-6115.
[8]Bachmann M,Mørøy T.Theserine/threoninekinasePim-1[J].The International Journal of Biochemistry & Cell Biology,2005,37(4):726-730.
[9]申海兰,赵靖松.分子动力学模拟方法概述[J].装备制造技术,2007,35(10):29-30+34.
[10]Nishiguchi G A,Atallah G,Bellamacina C,et al.Discoveryofnovel3,5-disubstitutedindolederivativesaspotentinhibitorsofPim-1,Pim-2,andPim-3proteinkinases[J].Bioorganic & Medicinal Chemistry Letters,2011,21(21):6366-6369.
[11]Wang J,Cieplak P,Kollman P A.Howwelldoesarestrainedelectrostaticpotential(RESP)modelperformincalculatingconformationalenergiesoforganicandbiologicalmolecules?[J].Journal of Computational Chemistry,2000,21(12):1049-1074.
[12]Wang J,Wolf R M,Caldwell J W,et al.Developmentandtestingofageneralamberforcefield[J].Journal of Computational Chemistry,2004,25(9):1157-1174.
[13]Jorgensen W L,Chandrasekhar J,Madura J D,et al.Comparisonofsimplepotentialfunctionsforsimulatingliquidwater[J].The Journal of Chemical Physics,1983,79(2):926-935.
[14]Ryckaert J P,Ciccotti G,Berendsen H J C.Numericalintegrationofthecartesianequationsofmotionofasystemwithconstraints:moleculardynamicsofn-alkanes[J].Journal of Computational Physics,1977,23(3):327-341.
[15]Kollman P A,Massova I,Reyes C,et al.Calculatingstructuresandfreeenergiesofcomplexmolecules:combiningmolecularmechanicsandcontinuummodels[J].Accounts of Chemical Research,2000,33(12):889-897.
[16]Bashford D,Case D A.Generalizedbornmodelsofmacromolecularsolvationeffects[J].Annual review of physical chemistry,2000,51(1):129-152.
[17]Weiser J,Shenkin P S,Still W C.Approximate atomic surfaces from linear combinations of pairwise overlaps(LCPO)[J].Journal of Computational Chemistry,1999,20(2):217-230.
[18]Feig M,Karanicolas J,Brooks C L.MMTSBToolSet:enhancedsamplingandmultiscalemodelingmethodsforapplicationsinstructuralbiology[J].Journal of Molecular Graphics and Modelling,2004,22(5):377-395.
[19]Daura X,Gademann K,Jaun B,et al.Peptidefolding:whensimulationmeetsexperiment[J].Angewandte Chemie International Edition,1999,38(1-2):236-240.
[20]Maisuradze G G,Liwo A,Scheraga H A.Relationbetweenfreeenergylandscapesofproteinsanddynamics[J].Journal of chemical theory and computation,2010,6(2):583-595.
[21]Hegger R,Altis A,Nguyen P H,et al.Howcomplexisthedynamicsofpeptidefolding?[J].Physical Review Letters,2007,98(2):028102.
[22]孙占莉,肖旭华,袁博.Pim-1激酶小分子抑制剂的研究进展[J].上海医药,2013,35(1):43-48+52.
[23]Hu J P,Gong X Q,Su J G,et al.StudyonthemolecularmechanismofinhibitingHIV-1integrasebyEBR28peptideviamolecularmodelingapproach[J].Biophysical Chemistry,2008,132(2):69-80.
Molecular Recognition Between Pim-1 Protein Kinase and 3,5-disubstituted Pyrazolopyridine Derivatives
LIANGLi1,HUJianping1,2,DUWenyi1,ZUOKe1,LIUWei1,GOUXiaojun1
(1.Key Laboratory of Medicinal and Edible Plants Resources Development, Chengdu University, Chengdu 610106, China;.College of Chemistry, Leshan Normal University, Leshan 614004, China)
Abstract:Pim-1 kinase,with serine/threonine kinase activity,can lead to tumor formation if it is over expressed,so it is a potential anti-cancer drug target.In this paper,a 20 ns molecular dynamics simulation is performed on the compound of PPK and a 3,5-disubstituted pyrazolopyridine derivative.The main driving force of the compound is analyzed from the perspective of energy.Then,the key function of I 185 in recognition is analyzed independently.Finally,the paper analyzes the conformational changes of the compound in the simulation process.The results show that the conservative property of the core region of PPK is an important mechanism for its physiological function.The simulation results are helpful for the following drug design of inhibitor based on PPK.
Key words:Pim-1 protein kinase;molecular dynamics;cluster analysis;free energy surface chart;conformational changes
文章编号:1004-5422(2016)02-0107-06
收稿日期:2016-03-02.
基金项目:国家自然科学基金(11247018,11147175)、 四川省教育厅科研重点课题(12ZA066)资助项目.
作者简介:梁立(1983 — ), 女, 硕士, 助理研究员, 从事生物信息学研究.
中图分类号:R914.5;R96
文献标志码:A
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
唐山师范学院学报(2020年6期)2020-04-16
温州大学学报(自然科学版)(2016年1期)2016-10-27
中学化学(2015年12期)2016-01-19
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
应用化工(2014年7期)2014-08-09
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年5期)2014-02-28
