
6例Gitelman综合征患者的临床及基因学分析
2016-07-22 02:44:06苗苗赵超群刘静李金慧王晓黎单忠艳
中国医科大学学报 2016年7期
关键词:基因突变
苗苗,赵超群,刘静,李金慧,王晓黎,单忠艳
(中国医科大学附属第一医院内分泌科,内分泌研究所,辽宁省内分泌疾病重点实验室,沈阳 110001)
6例Gitelman综合征患者的临床及基因学分析
苗苗,赵超群,刘静,李金慧,王晓黎,单忠艳
(中国医科大学附属第一医院内分泌科,内分泌研究所,辽宁省内分泌疾病重点实验室,沈阳 110001)
摘要在低钾血症患者中筛选出6例临床诊断符合Gitelman综合征(GS)的患者,分析其临床资料,对符合临床诊断患者的致病基因SLC12A3及经典型Bartter综合征(BS)的致病基因CLCNKB进行直接测序,寻找致病突变位点,探讨基因型与表型的联系。基因测序发现其中3例患者存在SLC12A3的致病突变。临床疑诊GS的患者需通过基因诊断确证,不携带基因变异的患者可能存在其他原因导致相似临床表型。
关键词Gitelman综合征;SLC12A3基因;CLCNKB基因;基因突变
网络出版地址
Gitelman综合征(Gitelman syndrome,GS)是一种常染色体隐性遗传的肾小管疾病,其临床表现与Bartter综合征(Bartter syndrome,BS)相似,主要临床特征为低钾血症、低镁血症、低尿钙、醛固酮水平增高但血压正常。GS在高加索人群中患病率大约为1/40 000,杂合子携带率高达1%,为最常见的遗传性肾小管疾病之一[1]。目前尚无对中国人GS发病率的数据统计。GS与位于16号染色体长臂上的SLC12A3基因突变有关[2]。SLC12A3基因编码噻嗪类利尿剂敏感的Na+/Cl-共同转运体(thiazide sensi⁃tive sodium chloride cotransporter,Na⁃Cl cotransport⁃er,NCCT),目前已发现超过140种SLC12A3基因的突变类型[3]。在少数的GS患者中发现编码氯通道的CLCNKB基因突变。
本研究对临床诊断为GS患者的病史、临床表现、实验室和影像学检查进行分析,并对其致病靶基因进行测序,探讨基因型与表型的联系。
1 材料与方法
1.1研究对象
选择2014年至2015年于中国医科大学第一附属医院内分泌科住院的低钾血症患者30例,根据GS的临床诊断标准[4]筛选出符合诊断标准的患者6例,其中男4例,女2例,年龄18~65岁,病程1~10年。
1.2诊断标准
低钾血症、碱中毒、高尿钾(>25 mmol/24 h)、低血镁(<0.66 mmol/L)、低尿钙肌酐比(<0.2),肾素血管紧张素活性升高但血压正常,且排除转移性低钾、胃肠道失钾、肾小管酸中毒,以及缓泄剂、利尿剂或乙醇的用药史[4]。
1.3病例简介
以下病例均排除其他低钾常见病因。
病例1:18岁男性,乏力等症状不明显,十余年来反复出现低血钾,经补钾对症治疗后低血钾仍未纠正。
病例2:22岁男性,四肢麻木、乏力、易摔倒2年,于医院就诊发现低钾,经对症补钾治疗后上述情况仍反复出现。
病例3:44岁男性,10余年发作性下肢无力病史,严重时蹲起困难,病情逐渐发展至频繁发作性肢体软瘫。
病例4:28岁女性,乏力、心悸病史5年,每次发病经对症治疗后好转,此次发病血钾最低达1.88 mmol/L,临床特点为发病年龄早,血压偏低。
病例5:55岁男性,下肢乏力及麻木病史10余年,发现血钾低9年,经对症补钾后仍不易纠正。临床特点为患者发病年龄较晚,同时伴有糖尿病及糖尿病周围神经病变。
病例6:65岁女性,周身乏力、气短伴恶心症状半年,化验发现血钾2.4 mmol/L,每次发作对症补钾后症状好转,临床特点为发病年龄晚,血压正常。
1.4检测方法
血电解质以全自动生化仪测定。促肾上腺皮质激素(adreno cortico tropic hormone,ACTH)、皮质醇采用免疫化学发光法测定,血肾素、血管紧张素、血醛固酮采用放射免疫法测定。
1.5基因检测
使用DNA提取试剂盒(天根生化科技北京有限公司)提取外周血基因组DNA,对PCR产物纯化(天根生化科技北京有限公司琼脂糖凝胶回收试剂盒),再对SLC12A3基因及CLCNKB基因进行全部外显子以及外显子-内含子交接部分的直接测序(上海新培晶检验所及北京六合华大基因科技股份有限公司),测序引物见以往文献[5]。
1.6应用在线软件预测突变的功能
应用在线蛋白质预测软件PolyPhen⁃2(http:// genetics.bwh.harvard.edu/pph2/)和SIFT(http://prove⁃an.jcvi.org/)对发现的突变位点进行功能预测。
2 结果
6例患者均存在高尿钾,低镁血症、低尿钙,肾素血管紧张素活性升高但血压正常,在排除转移性低钾、胃肠道失钾、肾小管酸中毒,以及致钾流失的用药史之后临床诊断GS,见表1。
基因诊断结果(表2,图1)提示3例患者存在导致SLC12A3基因功能减低的突变(L849H,T60M和L671P)。应用在线蛋白质功能预测软件PolyPhen⁃2和SIFT对此3处突变进行预测提示均会导致蛋白质的功能减低,见表3。
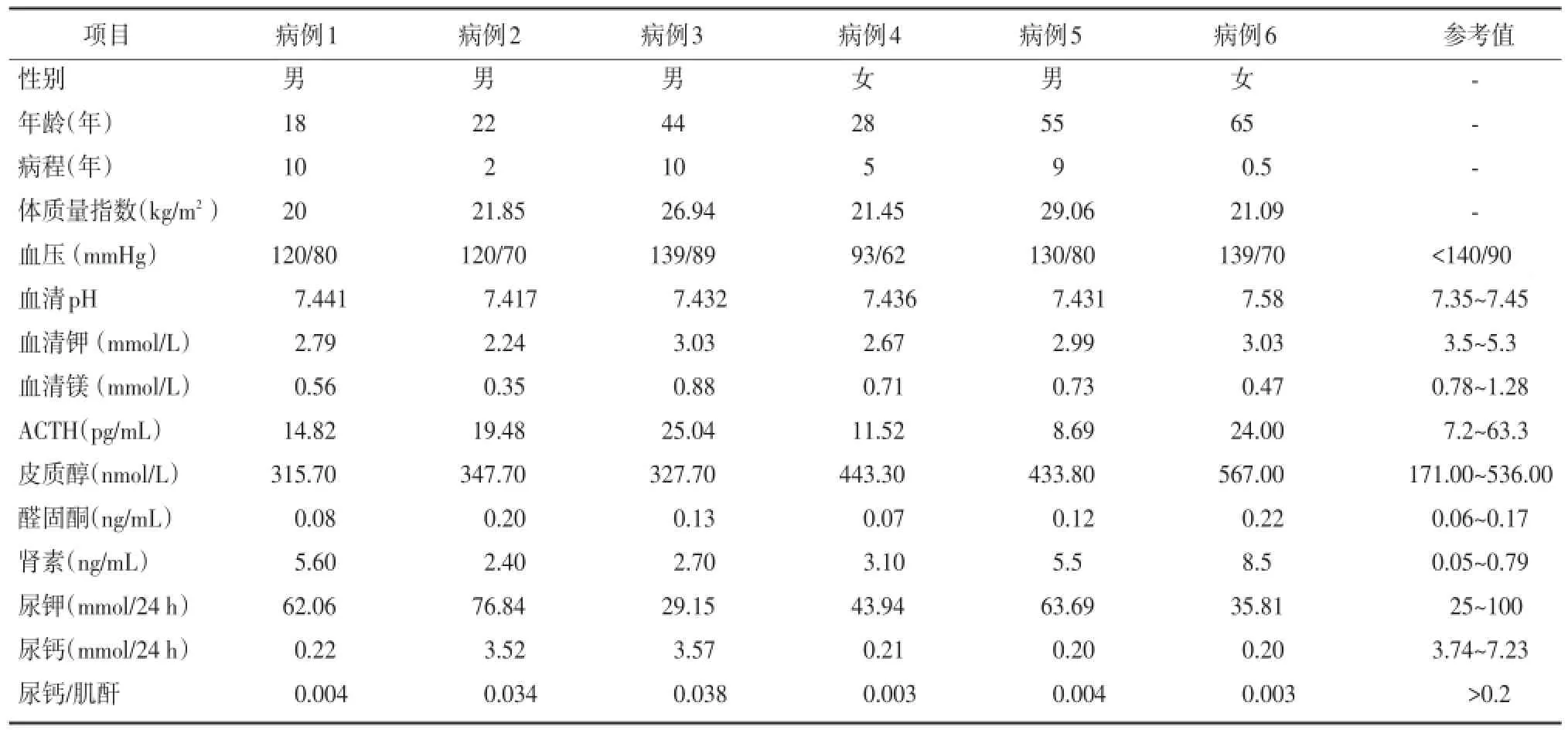
表1 病例基本资料及实验室检查结果

表2 6例基因检测结果
3 讨论
Gitelman[6]在1966年首次报道了3例22~47岁的女性患者,临床表现类似BS,但同时伴有低血镁、低尿钙,后被称为GS[7]。GS发病与位于16号染色体长臂上的SLC12A3基因突变有关[8-9],目前已发现140多个SLC12A3基因突变位点(人类基因突变数据库HGMD),包括错义突变、剪切突变、无义突变、读码框位移突变等,其中大部分以错义突变为主,复合杂合突变多于纯合子突变,无热点突变发现。国内对一组临床诊断为GS的患者进行基因学检测,结果发现T60M可能是中国患病人群最常见的突变位点[10],但并非所有的GS患者均携带SLC12A3基因突变,提示可能有其他遗传位点表现为相似的表型[11]。

图1 SLC12A3基因检测结果

表3 SLC12A3基因突变预测结果
本研究中6例患者均为近期于我科就诊的低钾血症患者,共同特点是存在低钾血症、低镁血症、肾性失钾但血压正常。因为BS与GS的临床表型部分重叠,因此本研究通过进一步对靶基因SLC12A3和CLCNKB的检测来明确是否存在导致GS的遗传学基础。3例患者发现了SLC12A3基因突变,其中患者2的突变类型为纯合型T60M,曾有报道[10]认为是中国人中GS患者特有的常见突变类型。患者1的突变类型为杂合型L849H,曾有文献[12-14]报道在日本GS患者中有相同基因型,并认为其仅存在于日本人种中。笔者认为L849H杂合突变不足以导致患者出现GS的临床表型,考虑可能存在基因调节区域的变异或者其他导致相似表型的基因中存在未知的变异。本研究中仅对导致经典型BS的靶基因进行了分析,未发现致病位点。另外,在日本人中的研究[15]表明携带L849H杂合突变的个体血钾与正常人相比差异具有统计学意义,甚至提出了“携带SLC12A3基因功能性杂合突变是影响血压水平的一个重要因素”的观点[16]。本研究中患者6的突变类型为杂合型L671P突变,目前尚未见国内外的文献报道,通过在线蛋白质功能预测软件预测,与T60M 和L849H同样位于蛋白质保守区域的L671P突变会损害蛋白质的正常功能,考虑可能为一处新的导致GS表型的基因突变类型。
此6例患者确诊后均接受补钾联合应用门冬氨酸钾镁和(或)安体舒通等治疗,低钾的临床症状(乏力、软瘫等)均得到缓解,但尚未完全纠正低血镁、低血钾,说明GS的离子紊乱不易纠正,需要多种药物联合长期应用。
GS是一种预后良好、进展缓慢的疾病,迄今报道的发展到终末期尿毒症的GS患者仅有2例[17-18]。但GS长期发展会影响患者生活质量,并有引起慢性肾功能不全的危险。基因诊断的意义在于早期确诊后进行针对性的治疗,本病尚无法根治,治疗应对症补钾、补镁,前列腺素合成酶抑制剂、醛固酮拮抗剂等多种药物联合应用。BS患者的前列腺素E2往往是升高的,而GS患者绝大多数前列腺素E2为正常,据此有研究[19]认为应用环氧化酶抑制剂治疗GS效果不佳。本研究中的6例病例提示临床工作中应注意顽固性低钾血症的患者是否存在GS,对高度怀疑GS的患者应尽早行基因诊断检测,从而提高对GS的认识和诊治水平,提高患者生活质量。
参考文献:
[1]KNOERS NV,LEVTCHENKO EN.Gitleman syndrom[J].Or⁃phanet J Rare Dis,2008,3:22.
[2]GALLI⁃TSINOPOULOU A,PATSEADOU M,HATZIDIMITRIOU A.Gitelman syndrome:first report of genetically established diagno⁃sis in Greece[J].Hippokratia,2010,14(1):42-44.
[3]SIMON DB,NELSON WC,BIA MJ.Gitelman variant of Bartter's syndrome,inherited hypokalaemic alkalosis,is caused by mutations in the thiazide⁃sensitive Na⁃Cl cotransporter[J].Nat Genet,1996,12(1):24-30.
[4]SINHA A,LNĚNIKA P,BASU B.Gitelman syndrome:novel muta⁃tion and long⁃term follow⁃up[J].J Clin Endocrinol Metab,2012,16 (2):306-309.
[5]MONNENS L,BINDELS R,GRUNFELD JP.Gitelman syndrome comes of age[J].Nephro1 Dial Transplant,1998,13(7):1617-1619.
[6]杨国庆,赵蕾,席文琪,等.Gitelman综合征9例临床分析[J].中华内科杂志,2006,45(8):650-653.
[7]LEMMINK HH,KNOERS NV,KÁROLYI L.Novel mutations in the thiazide⁃sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C⁃terminal domain [J].Kidney Int,1998,54(3):720-730.
[8]GITELMAN HJ,GRAHAM JB,WELT LG.A new familial disorder characterized by hypokalemia and hypomagnesemia[J].Trans As⁃soc Am Physicians,1966,79(3):221-235.
[9]GALLI⁃TSINOPOULOU A,PATSEADOU M,HATZIDI MITRIOU A.Gitelman syndrome:first report of genetically established diagno⁃sis in Greece[J].Hippokratia,2010,14(1):42.
[10]邵乐平,任红,王伟铭.Gitelman综合征SLC12A3基因突变研究[J].中华肾脏病杂志,2007,66(23):351-356.
[11]QIN L,SHAO L,REN H.Identification of five novel variants in the thiazide⁃sensitive NaCl co⁃transporter gene in Chinese pa⁃tients with Gitelman syndrome[J].Nephrology(Carlton),2009,14(1):52-58.
[12]MONKAWA T,KURIHARA I,KOBAYASHI K.Novel mutations in thiazide⁃sensitive Na⁃Cl cotransporter gene of patients with Gitelman's syndrome[J].J Am Nephrol,2000,11(1):65-70.
[13]MAKI N,KOMATSUDA A,WAKUI H.Four novel mutations in the thiazide⁃sensitive Na⁃Cl co⁃transporter gene in Japanese pa⁃tients with Gitelman's syndrome[J].Nephrol Dial Transplant,2004,19(7):1761-1766.
[14]AOI N,NAKAYAMA T,TAHIRA Y,et al.Two novel genotypes of the thiazide⁃sensitive Na⁃Cl cotransporter(SLC12A3)gene in pa⁃tients with Gitelman's syndrome[J].Endocrine,2007,31(2):149-153.
[15]NARABA H,KOKUBO Y,TOMOIKE H,et al.Functional confir⁃mation of Gitelman's Syndrome mutations in Japanese[J].Hyper⁃tens Res,2005,28(10):805-809.
[16]TAGO N,KOKUBO Y,INAMOTO N,et al.A high prevalence of Gitelman syndrome mutations in Japanese[J].Hypertens Res,2004,27(5):327-331.
[17]BONFANTE L,DAVIS PA,SPINELLO M,et al.Chronic renal fail⁃ure,end⁃stage renal disease,and peritoneal dialysis in Gitelman syndrome[J].Am J Kidney Dis,2001,38(1):165-168.
[18]CALÒ LA,MARCHINI F,DAVIS PA,et al.Kidney transplant in Gitelman's syndrome:report of the first case[J].Nephrol,2003,16(1):144-147.
[19]KURTZ I.Molecular pathogenesis of Bartter's and Gitelman[J]. Kidney Int,1998,54(4):1396-1410.
(编辑于溪)
网络出版时间:
中图分类号R589.9
文献标志码A
文章编号0258-4646(2016)07-0649-04
DOI:10.12007/j.issn.0258⁃4646.2016.07.016
基金项目:国家自然基金青年基金(81200653);卫生部国家临床重点专科资助
作者简介:苗苗(1989-),女,硕士研究生.
通信作者:王晓黎,E-mail:wlittlepear@163.com
收稿日期:2015-10-23
AClinicalandGeneticAnalysisof6CasesofGitelmanSyndrome
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国医学影像学杂志(2021年6期)2021-08-13 08:43:36
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
中国生殖健康(2018年2期)2018-01-12 13:57:51
现代检验医学杂志(2016年4期)2016-11-15 02:01:14
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
中华皮肤科杂志(2014年4期)2014-12-19 12:55:49
中国神经精神疾病杂志(2014年1期)2014-03-01 03:23:22
