
YC分子基态的结构与势能曲线* 1
2016-07-18 05:55:59彭伟成许永强
赣南师范大学学报 2016年3期
彭伟成,许永强
(赣南师范学院 a.新闻与传播学院;b.物理与电子信息学院,江西 赣州 341000)
YC分子基态的结构与势能曲线* 1
彭伟成a,许永强b
(赣南师范学院 a.新闻与传播学院;b.物理与电子信息学院,江西 赣州341000)
摘要:考虑到研究对象的相对论效应和电子相关效应,对于YC双原子分子,对Y采用赝势基组,C原子选用6-311+G(3df)和AUG-cc-PVTZ基组,计算方法使用Becke的交换泛函和三参数混合泛函形式即B3LYP杂化泛函方法,首先对YC双原子分子的最优结构进行了计算,由此得到分子的稳定构型、最低能量和振动频率.基于原子分子反应静力学原理,推导得到YC双原子分子基态的合理离解极限.通过对比以往文献报道的实验和高水平理论计算结果,发现CRENBL ECP/6-311+G(3df)混合基组为对体系进行计算最为合适.因此,就在B3LYP/ CRENBL ECP/6-311+G(3df)理论水平进一步对YC双原子分子基态的势能面进行了刚性扫描.基于扫描结果,并采用最小二乘法拟合,得到了10参数的Murrell-Sorbie势能函数曲线.由曲线系数(De,a1,a2,a3,a4)进一步计算得到了二、三、四阶力常数(f2,f3,f4)以及相关光谱数据(Be, αe, ωe, ωeχe,De).为原子分子碰撞研究提供了有效的数据支持.
关键词:YC;基态;结构;势能曲线;光谱数据
1引言
在有气态碳参与的工业生产和应用过程中,过渡金属及其合金经常会作为重要的催化剂而参与这些化学反应过程,碳与催化剂之间会发生一些副反应并相应有副产物出现.这些副产物,如过渡金属碳化物,也引起了相当大的科学和技术兴趣. 钇(Y)是重要的过渡金属, 其外层电子组态是4d15s2,d轨道只有1个电子故较为活跃,它的金属活泼性仅次于碱金属和碱土金属,并可以和碳等发生反应.YC双原子分子作为最简单的钇碳化合物,当用Y或者其合金作为催化剂时,YC是极易得到的.为了区分正副反应及其产物特性,对YC的研究也是必不可少的.然而,据我们所知,对YC进行理论和实验研究的结果还非常有限.一是Shim等[1]通过多组态自洽场计算和质谱实验报道了YC分子的低电子态组成和化学键的特征,并得到了其离解能的值.二是Simard等[2]通过光谱实验进一步证明了Shim等的研究成果,即YC的基态为4П态,部分激发态的特征在此也进行了讨论.因此, 目前对于YC的研究还非常有限.这里我们主要关注的是YC基态势能函数曲线的性质.势能函数是研究原子分子碰撞和分子反应动力学的基础,也是研究分子稳定性的依据.它在辐射化学、激光化学等方面有广泛应用[3-9].另外,YC的势能函数的研究,对于研究更复杂的钇碳三原子及其更复杂结构的构型、频率和化学反应等都有理论上的帮助.因此,研究具有重要的理论意义和实际应用价值.
2计算方法
由群理论,基于广义Wigner-Witmer规则,运用原子分子反应静力学理论中的分离原子方法[10-11],可以确定相关的电子态.Y和C的基态电子状态的原子群的不可约表示分别为2Dg和3Pg,均属于SU(n)群,当两个原子靠近时,对称性降低,生成的YC双原子分子属于C∞v群.把SU(n)群的不可约表示分解为C∞v群的不可约表示的直和,通过直积和约化可得分子的电子态.Y(2Dg)分解为C∞v群的不可约表示的直和为2Dg=2∑+⊕2П⊕2Δ,C(3Pg)分解为C∞v群的不可约表示的直和为3Pg=3∑-⊕3П.两者进一步直积和约化,结果为
(3∑-⊕3П)⊕(2∑+⊕2П⊕2Δ)=2,4∑+⊕2,4∑-(2)⊕2,4П(3)⊕2,4Δ(2)⊕2,4Φ
结合以往文献报道和优化计算结果可知,YC分子基态为4П态.而从以上直积约化结果来看,恰好含有YC分子的基态4П态,由此可知,从Y(2Dg)和C(3Pg)基态原子群的不可约表示组合可以得到YC分子的基电子态.由微观过程的可逆性原理,可得YC双原子分子的基电子态的离解极限为
YC (X4П)→Y(2Dg)+C(3Pg)
由原子物理学和物质结构理论知识可知,Y在周期表中第五排,核外电子数为39,轨道电子排布为 1s22s22p63s23p63d104s24p64d15s2.考虑到钇属于长周期中的过渡金属元素,电子数较多和具有相对论效应的实际情况,因此对钇的所有计算都是采用相对论赝势进行.这里把核外电子分成两部分来考虑,其中中心电子数28个(1s22s22p63s23p63d10),价电子数11个(4s24p64d15s2),对Y分别采用LANL2DZ,LANL2TZ,Stuttgart RSC 1997 ECP,CRENBL ECP赝势基组,对C采用6-311+G(3df),AUG-cc-PVTZ两种全电子基函数.在Gaussian09程序包中应用Becke的交换泛函和三参数混合泛函形式即B3LYP杂化泛函计算方法,对YC基态构型进行了优化和频率计算.通过理论分析结果和已有的实验数据和高水平理论计算结果对比分析发现,所采用基组的混合组合CRENBL ECP/6-311+G(3df)对YC分子进行计算的最合适.因此,进一步在B3LYP/ CRENBL ECP/6-311+G(3df)理论水平对YC分子基态进行了单点能刚性扫描,所有结果概括于表1中.这里之所以采用B3LYP杂化泛函,是由于这种泛函形式既充分考虑了电子相关效应,又拥有比较高的计算效率,因此,该方法在量子化学和第一性原理计算方面也是被广泛采用的.
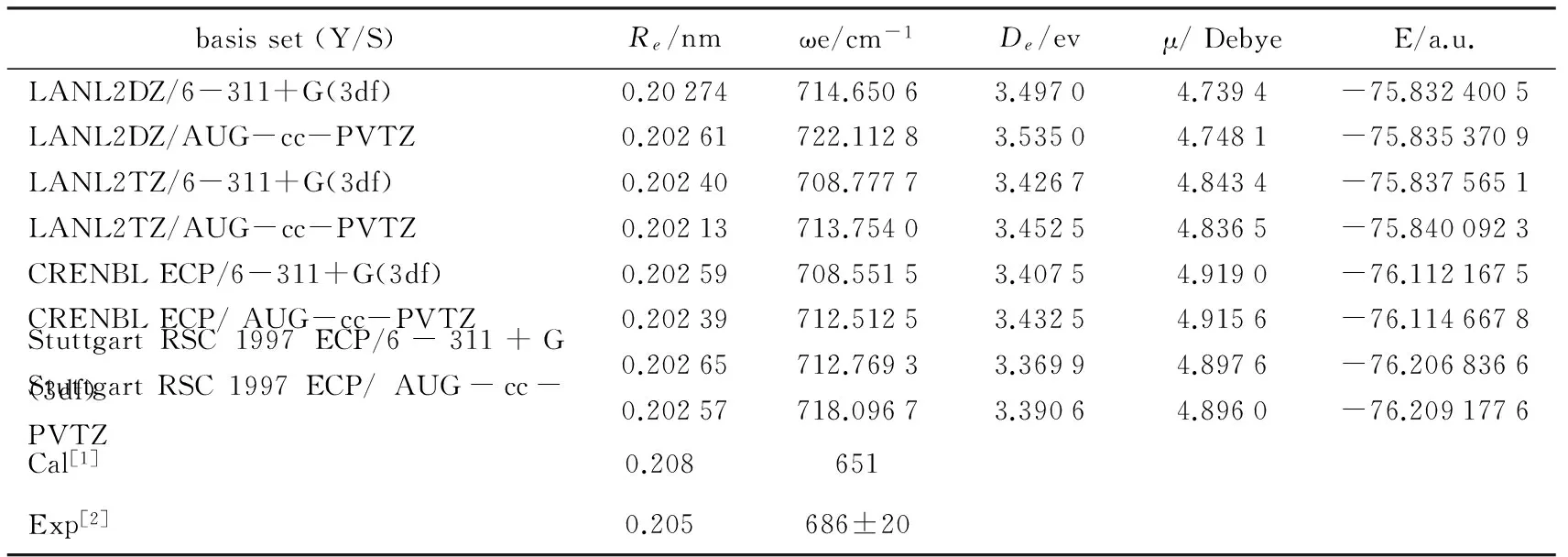
表1 YC分子基态的优化计算结果与已有实验和理论值的比较
对于基态双原子分子,Murrell-Sorbie(MS)和Duham势能函数均是很好的解析势能函数形式.它们不但能较准确反映平衡位置附近的排斥支和吸引支情况,且具有较好的长程势.因此,这里我们采用了MS势能函数来进行曲线拟合.从以往经验来看,对于轻元素所形成的双原子分子,一般采用4参数的MS函数就能得到比较满意的结果,而在这里Y是过渡金属,研究发现采用10参数的MS函数更符合实际情况,即采用以下具体势能函数形式
(1)
式中ρ=r-Re,r是核间距离,Re为两原子之间的平衡距离.De,ai(i=1,2,3,…,9)为势能函数曲线形式的拟合系数,且De具有物理意义,即为分子的离解能.根据以上势能曲线形式进一步求其二阶、三阶和四阶导数,可得力常数和势能函数曲线系数的关系如下:
(2)
(3)
(4)
f2,f3,f4分别为二阶,三阶和四阶力常数,根据力常数结果和平衡距离Re,进一步可以得到光谱常数(Be, αe, ωe, ωeχe)[11]:
(5)
(6)
(7)
(8)
式中,Be为平衡转动常数,αe为振动-转动耦合常数,ωe为谐振频率,ωeχe为非谐性频率,u为约化质量,c为真空中的光速.
3结果和讨论
基于表1中所例杂化泛函在各种不同混合基组下所得计算结果,再综合谐性频率ωe和平衡核距离Re的理论和实验报道结果来看,在使用密度泛函理论的B3LYP方法的情况下,采用CRENBL ECP/6-311+G(3df)混合基组对YC双原子分子进行计算是较合适的.基于此,进一步在B3LYP/CRENBL ECP/6-311+G(3df)理论水平对YC基态构型进行了势能面刚性扫描计算.扫描范围为0.12-0.92 nm,步长为0.001 nm,共计算了800个单点能值.利用这些单点势能值,再采用最小二乘法拟合到(1)式.由拟合所得MS函数系数De,a1,a2,a3,a4,运用(2)-(4)式计算得到二、三、四阶力常数f2,f3,f4.所有结果收集于表2中.

表2 YC分子基态的Murrell-Sorbie势能函数和力常数
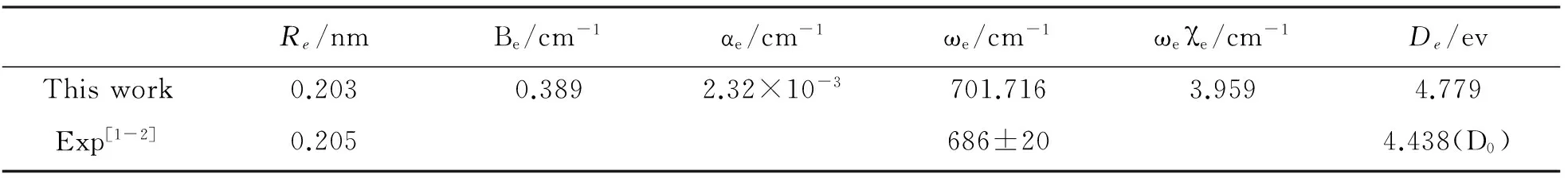
表3 YC分子基态的光谱常数
根据(5)-(8)的关系,进而得到YC分子基态的光谱数据Be,αe,ωe,ωeχe,所得结果如表3所示.从表3可知,文献[2]通过光谱实验测量报道的平衡键长Re和谐性频率ωe的值分别为0.205 nm和686±20 cm-1,这与我们的计算结果0.203 nm和701.716 cm-1是非常吻合的.其次,从离解能的结果来看,我们的计算结果为4.779 ev,而文献[1]报道的离解能为4.438 ev,我们的结果要略大于文献[1]的值,这其实是合理的,原因在于文献[1]报道的离解能为没有包含零点能修正的结果,如果加上零点能修正值,那么两者的值将会进一步接近.由此可知,势能面扫描结果是与实际情况比较吻合的,因此,在此基础上所得到的其它光谱数据也是可信的.

图1 YC分子基态的势能曲线
图1为YC双原子分子基态的势能曲线,图中的圆圈为单点势能值,实线为势能曲线拟合结果.从图1来可以看出,无论是在平衡位置附近,还是在吸引支和排斥支的远程位置,扫描得到的单点能结果与拟合曲线的拟合值均较符合,这说明10参数的MS势能函数形式能正确反映YC分子基态的势能曲线.
4结论
考虑到研究对象的电子相关效应和相对论效应,对Y分别选用LANL2DZ,LANL2TZ,Stuttgart RSC 1997 ECP,CRENBL ECP相对论赝势基组,对C分别选用6-311+G(3df)、AUG-cc-PVTZ基组,运用Becke的交换泛函和三参数混合泛函形式即B3LYP杂化泛函方法,对YC分子基态进行了结构优化,得到了基态的稳定构型和构型的最低能量.运用群理论和原子分子反应静力学原理中的分离原子方法,结合微观过程的可逆性原理,得到了YC双原子分子基态的离解极限.进一步,在杂化泛函B3LYP方法下,采用CRENBL ECP/6-311+G(3df)混合基组,对YC基态的势能面进行了单点能扫描.然后利用扫描结果,最小二乘拟合得到了分子基态的MS势能曲线.基于势能曲线系数和力常数计算得到了相关的光谱数据.对比已有的实验数据发现,两者吻合很好.这为原子分子碰撞和分子动力学的研究提供了很好的数据支持.
参考文献:
[1]Shim I, Pelino M, Gingerich K A. Electronic states and nature of bonding in the molecule YC by all electron ab initio multiconfiguration self-consistent-field calculations and mass spectrometric equilibrium experiments[J].J. Chem. Phys. 1992, 97:9240.
[2]Simard B, Hackett P A, Balfour W J. Jet-cooled optical spectroscopy of yttrium monocarbide, evidence for a4Пiground state[J].Chem. phys. Lett. 1994, 230:103.
[3]Guan Y, Han X, Yang J, et al. Updated potential energy function of the Rb2a3∑+state in the attractive and repulsive regions determined from its joint analysis with the 23П0gstate[J].J. Chem. Phys. 2013, 139:144303.
[4]Koput J. Ab initio potential energy surface and vibration-rotation energy levels of lithium monohydroxide[J].J. Chem. Phys. 2013, 138:234301.
[5]Hajigeorgiou P G. The potential energy function of the ground electronic state of16O2[J].J. Chem. Phys. 2013,138:014309.
[6]Legon A C. A reduced radial potential energy function for the halogen bond and the hydrogen bond in complexes B...XY and B...HX, where X and Y are halogen atoms[J].Phys. Chem. Chem. Phys. 2014,16:12415.
[7]Wu D L, Tan B, Wan H J, et al. Molecular properties and potential energy function model of BH under external electric field[J].Chin. Phys. B. 2013,22:123101.
[8]Jalili A H, Behnejad H, Afsahi G, et al. Potential energy function for HeS+ and transport properties of S+in He[J].Chem. Phys. Lett. 2013,584:49.
[9]Zhang L L, Gao S B, Meng Q T, et al. Accurate ab initio-based analytical potential energy function for S2(a1Δg) via extrapolation to the complete basis set limit[J].Chin Phys. B. 2015,24:013101.
[10]朱正和.原子分子反应静力学[M].北京:科学出版社,1997:23-27.
[11]朱正和,俞华根.分子结构与分子势能函数[M].北京:科学出版社,1997:56-62.
* 收稿日期:2016-01-03
DOI:10.13698/j.cnki.cn36-1037/c.2016.03.007
基金项目:江西省科技厅青年基金项目(2014BAB216011);赣南师范学院自然科学研究课题(430374)
作者简介:彭伟成(1979-),女,湖南娄底人,赣南师范学院新闻与传播学院讲师,研究方向:信息技术理论;许永强(1978-),男,湖南邵阳人,赣南师范学院物理与电子信息学院讲师,研究方向:光谱的实验和理论.
中图分类号:O561.1
文献标志码:A
文章编号:1004-8332(2016)03-0026-04
Structure and Potential Energy Curve of the Ground State of YC Molecule
PENG Weicheng, XU Yongqiang
(SchoolofPhysicsandElectronicInformation,GannanNormalUniversity,Ganzhou341000,China)
Abstract:In view of the relativistic effect and the electron correlation effect for YC diatomic molecule, using pseudo potential basis set for Y atom and 6-311+G(3df) as well as AUG-cc-PVTZ basis set for C atom, the optimal structure of YC diatomic molecule is calculated using Becke's nonlocal three-parameter exchange and correlation functional with the Lee-Yang-Parr correlation functional, B3LYP. And the stable geometry, ground state's energy, along with harmonic frequency are obtained. Based on the theory of atomic and molecular statics, the reasonable dissociation limit of the ground state of YC diatomic molecule is derived. By comparing the results of calculations with the existing experimental and high accuracy theoretical calculation values that literature reported, it can concluded that the mixed basis sets CRENBL ECP/6-311+G(3df) is the most suitable for the calculation of the molecule. Accordingly, the potential energy surfaces of ground state for YC diatomic molecule has been rigid scanned at the B3LYP/ CRENBL ECP/6-311+G (3df) level. On account of the results of scanning, Murrell-Sorbie potential energy function curve with ten parameters is gained by least square fitting. The quadratic, cubic and quartic force contants (f2,f3,f4)as well as spectroscopic data (Be, αe, ωe, ωeχe,De) are calculated according to curve's coefficients(De,a1,a2,a3,a4). The studies provide effective data support for the research of atomic and molecule collision.
Key words:YC; ground state; structure; potential energy curve; spectroscopic data
网络出版地址:http://www.cnki.net/kcms/detail/36.1037.C.20160510.1219.042.html
猜你喜欢
数学物理学报(2022年3期)2022-05-25 13:33:22
数学物理学报(2022年2期)2022-04-26 14:08:10
数学物理学报(2022年1期)2022-03-16 06:15:04
数学物理学报(2021年5期)2021-11-19 07:01:16
哲学评论(2021年2期)2021-08-22 01:53:34
数学物理学报(2021年3期)2021-07-19 06:02:18
中华诗词(2019年7期)2019-11-25 01:43:04
数学物理学报(2019年3期)2019-07-23 01:15:24
模具制造(2019年3期)2019-06-06 02:10:54
影视与戏剧评论(2016年0期)2016-11-23 05:26:01
