
载体碱性对Fe基催化剂费-托合成反应的影响
2016-06-24 06:49张俊张征湃苏俊杰付东龙戴薇薇刘达徐晶韩一帆华东理工大学化学工程联合国家重点实验室上海200237
化工学报 2016年2期
张俊,张征湃,苏俊杰,付东龙,戴薇薇,刘达,徐晶,韩一帆(华东理工大学化学工程联合国家重点实验室,上海 200237)
载体碱性对Fe基催化剂费-托合成反应的影响
张俊,张征湃,苏俊杰,付东龙,戴薇薇,刘达,徐晶,韩一帆
(华东理工大学化学工程联合国家重点实验室,上海 200237)
摘要:低碳烃类化合物是化学工业中重要的有机原料,通过非石油路线由费-托反应(Fischer-Tropsch)制备低碳烃类具有巨大前景,载体对于费-托合成催化剂的反应产物分布具有重要影响。探究了载体碱性对负载型Fe基催化剂在费-托合成反应中反应性能的影响。通过浸渍法制备了Fe20/AlPO4、Fe20/γ-Al2O3、Fe20/MgAl2O4催化剂,考评结果表明,载体碱性越强,碳链增长概率(α值)越大,C5+选择性上升,烯烷比(O/P)增加。通过Raman 光谱和TPH实验对由柠檬酸铁铵为前体煅烧后的催化剂表层碳物种进行分析表明,载体碱性越强,催化剂表面碳石墨化程度越高,吸附碳数量越少。并依托XRD、H2-TPR、CO2-TPR表征信息构建了不同碱性载体负载的Fe基催化剂的构效关系,表明载体给电子能力的强弱引起催化剂表面碳物质含量和金属载体相互作用的差异,最终导致了催化活性和选择性的不同。
关键词:费-托合成;合成气;催化;Fe基催化剂;催化剂载体;碱性;碳物种
2015-08-03收到初稿,2015-10-13收到修改稿。
联系人:韩一帆。第一作者:张俊(1993—),男,硕士研究生。
Received date: 2015-08-03.
引 言
低碳烃类化合物是化学工业中重要的有机原料,广泛应用于塑料、溶剂、制药等领域。传统的低碳烃类产品主要来源于石脑油的热裂解或催化裂解[1]、烷烃的脱氢[2-3]。然而,迫于环境和能源安全的压力,开辟新型非石油路线制备低碳化学产品的工艺变得更为迫切。
近年来,以煤为原料制备高附加值化工产品得到快速发展,一大批新型煤化工技术正走向产业化[4]。同时,以生物质为原料生产化工产品的技术路线也越来越受到人们的重视。生物质或生物油制备化工产品也取得了一定进展[5]。而合成气(H2+CO)作为一种独特的化工原料,来源广泛。煤气化及生物质气化技术均可以规模化生产合成气。基于煤化工及生物化工的背景,以合成气为原料制备高附加值化工产品已经成为当下的热门研究课题。
目前,通过合成气制备低碳烃类化合物主要有两类工艺。一类是间接法,通常由至少两步工艺组成:①合成气制备甲醇,甲醇再制备烃类产品[6];②费-托合成制油,合成油再裂解制备低碳烃类[7]。另一类是直接法,即费-托合成直接制备低碳化合物路径。后者作为一种直接法工艺,没有中间步骤,相比之下具有更高的经济效益和工艺合理性[8]。
多年来,由于Fe基催化剂价廉易得、对原料气要求低、抗毒性好等优点,开发新型Fe 基催化剂通过费-托合成直接制备低碳烃类产品受到广泛关注[9]。而负载型催化剂不仅可以提升催化剂的机械强度,并可以提高活性金属的分散性,为活性金属提供适宜的配位环境[10]。但受到多种因素的影响,负载型催化剂的费-托反应活性及烃类选择性往往较低[11-14]。
为进一步研究载体对Fe基费-托合成催化剂的作用,本工作着眼于载体碱性对Fe基催化剂费-托合成反应性能的影响,选用AlPO4、γ-Al2O3和MgAl2O43种载体,采用等体积浸渍法制备了一系列Fe基催化剂,通过催化剂活性评价及X 射线衍射、Raman光谱、程序升温吸附等表征技术探究了载体碱性在Fe基催化剂中对费-托合成反应性能的影响,并建立了相应的构效关系,为负载型Fe 基费-托反应催化剂的进一步理性设计提供参考。
1 实验部分
1.1载体材料的制备
柠檬酸铁铵(C6H11FeNO7,≥99.0%)及氧化铝(γ-Al2O3,≥99.0%)均由上海晶纯生化科技股份有限公司提供。镁铝尖晶石(MgAl2O4)的制备方法已有报道[15],其主要步骤如下:139 g Mg(NO3)2·6H2O和34 g Al(NO3)3·9H2O 按摩尔比6:1溶解在1000 ml去离子水中,形成溶液A;4.8 g Na2CO3和45.3 g NaOH 溶解在250 ml去离子水中,形成溶液B。使用自动进样泵将溶液B以2 ml·min−1的速度滴入溶液A中,混合液在333 K条件下搅拌1 h,并在室温下老化18 h。之后,使用2 μm滤膜过滤得到的固体在383 K下干燥12 h,最后在马弗炉中以2 K·min−1的升温速率升温至973 K,煅烧10 h。磷酸铝(AlPO4)参照前期文献报道的方法制备[16],其主要步骤如下:1.0 mol·L−1H3PO4溶液逐滴滴入到1.0 mol·L−1Al(NO3)3溶液中,并不断搅拌,之后使用氨水将其上层液体的pH调至4.5,所得到的白色凝胶经过洗涤、干燥后,在马弗炉中于1273 K煅烧5 h。
1.2催化剂的制备
在Fe基费-托合成反应中,催化剂上的碳物种信息丰富,而且对反应有着重要影响[17]。其碳物种可来源于催化剂制备及反应过程。本实验使用C6H11FeNO7为前体,在催化剂制备过程中引入碳源,使用等量浸渍法制备20.0%(质量分数)Fe 负载在不同载体上的催化剂。制备方法如下:分别将含Fe 0.20 g的C6H11FeNO7颗粒加入一定量的去离子水中溶解,溶液缓慢滴入0.80 g不同载体中,搅拌均匀。用于溶解的去离子水用量依据不同载体的饱和吸水率确定。经298 K放置4 h后,于383 K干燥12 h,然后在100 ml·min−1氮气下于773 K煅烧5 h,氮气气氛中降温至室温后,逐步提高气氛中氧气含量进行钝化,直至氧气体积分数为20%,然后密封保存。
1.3催化剂活性评价
本实验使用高温高压微型反应催化评价装置进行催化性能测试。评价系统由反应器、预热器、气相色谱等构成,实验装置如图1所示。催化剂首先在微型反应器内进行常压原位还原。还原气氛为H2(纯度为99.999%),流量为30 ml·min−1,以5 K·min−1的升温速率升温至623 K,保持5 h还原。
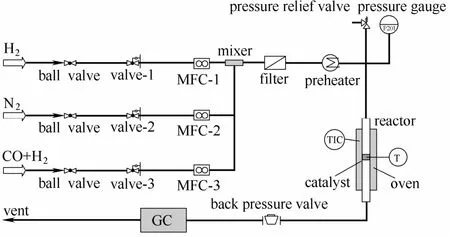
图1 催化剂评价装置Fig.1 Flowchart of experimental apparatus
催化剂活性考评在533 K、2.0 MPa 下进行。原料气为H2:CO=1:1(体积比)的混合气,流量为30 ml·min−1。催化剂装填量为100 mg,使用165~198 μm的SiO2进行稀释,稀释质量比为1:1。混合气由气体钢瓶经减压阀及预热器后进入反应器内,气相产物经减压阀后进入气相色谱系统进行在线采样检测,通过色谱峰面积及校正因子计算相应产物。其中H2和CO通过5A分子筛后接热导池检测器(TCD)检测;CO2通过TDX-01毛细管柱分离,经甲烷化炉后通过氢火焰检测器(FID 1)检测;烃类产物由HP Plot-Q毛细管柱后接氢火焰检测器(FID 2)检测。
1.4催化剂表征
XRD在布鲁克AXS有限公司的X射线多晶衍射仪上进行,采用波长0.154056 nm的CuKα线,管电压为40 kV,管电流为100 mA。扫描步长为0.02°,扫描速率为12(°)·min−1。
氮气低温吸附在美国Micromeritics的全自动物化吸附分析仪(仪器型号ASAP2010C)上进行。催化剂样品在真空状态下以523 K脱气处理6 h,随后在77 K下进行N2吸附。
氢气程序升温还原(H2-TPR)实验在10% H2气氛下(流量为30 ml·min−1)进行,以10 K·min−1的升温速率由303 K升温至1073 K还原,通过热导池检测器(TCD)检测出口尾气的H2浓度。
CO2-TPD实验在Linkam反应池中进行,样品装填量为50 mg。样品在423 K使用Ar(99.999%)吹扫2 h以去除表面吸附的水和杂质,Ar流量为30 ml·min−1。催化剂的CO2-TPD在吸附前先使用10% H2进行原位还原。还原条件:H2流量为30 ml·min−1,在623 K还原5 h。前处理后,在303 K通入CO2(99.999%)吹扫1 h进行吸附,流量为20 ml·min−1。吸附后使用Ar吹扫物理吸附的气体分子,直到基线平稳。TPD的升温速率为10 K·min−1,由303 K升温至1073 K。
程序升温加氢(TPH)实验同样在Linkam反应池中进行,样品装填量为50 mg。样品前处理与CO2-TPD类似,处理后的催化剂冷却至室温,将气氛切为10% H2/Ar下以15 K·min−1的升温速率升温至1023 K,同时用质谱仪检测尾气中荷质比为16 (CH4)的组分。
Raman光谱实验在Horiba公司的LabRAM HR拉曼光谱仪上进行,所用激光光源为514 nm Ar+离子激光器。显微系统为耦合在光谱仪上的Olympus显微镜,测试时采用50倍长焦物镜。采用针孔共焦技术,共焦针孔均采用300 µm。峰位采用单晶硅一阶峰(520.7 cm−1)矫正。散射信号通过180°背散射方式采集后到达CCD检测器成像,经过计算机处理得到样品的拉曼光谱。
1.5反应产物的定量分析
CO转化率XCO的计算公式如式(1)所示
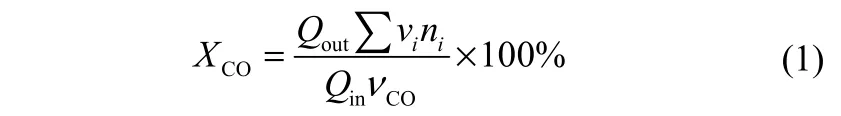
组分i的选择性Si的计算公式如式(2)所示

STY的计算公式如式(3)所示
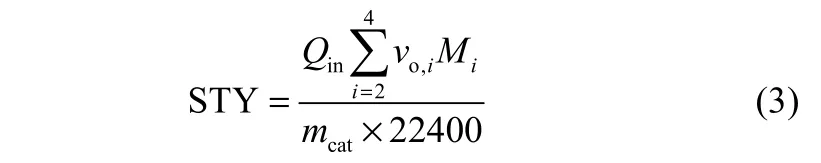
式中,Qout和Qin分别表示反应器出口和进口的气流量;vi表示出口气体中各组分的体积分数;vCO表示原料气中CO的体积分数;ni表示出口气体中各组分的碳数;vo,i表示出口气体中各低碳烯烃的体积分数;Mi表示出口气体中各低碳烯烃的摩尔质量,g·mol−1;mcat表示催化剂装填量,kg。
Herington[18]假设链增长概率为常数,最早提出了费-托合成反应机理。在此基础上Anderson等[19]提出了著名的费-托合成产物分布公式,即Anderson-Schulz-Flory(ASF)方程
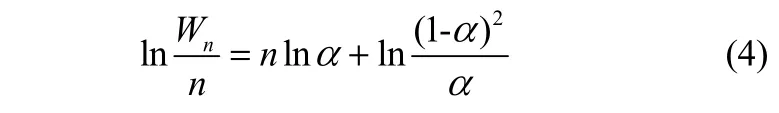
式中,α为链增长概率,Wn表示碳数为n的组分的质量分数。
2 实验结果与讨论
2.1载体碱性比较
通过CO2-TPD实验考察了3种载体的碱性(图2)。由图2可知,室温下AlPO4对CO2几乎没有吸附;γ-Al2O3在383K下存在一个较弱的吸附峰;MgAl2O4在403 K下存在强吸附峰。由脱附温度和脱附面积可知,AlPO4、γ-Al2O3、MgAl2O4碱性和碱性位数量依次增强。

图2 AlPO4、γ-Al2O3和MgAl2O4的CO2-TPD谱图Fig.2 CO2-TPD profiles of AlPO4, γ-Al2O3and MgAl2O4
2.2催化剂反应性能比较
Fe20/AlPO4(SBET= 77.4 m2·g−1,Vtotal= 0.30 cm3·g−1)、Fe20/γ-Al2O3(SBET= 120.2 m2·g−1,Vtotal= 0.34 cm3·g−1)和Fe20/MgAl2O4(SBET=150.2 m2·g−1,Vtotal= 0.20 cm3·g−1)催化剂在高温高压微型反应器上进行费-托催化性能测试。对Fe基催化剂费-托合成反应而言,活性中心一直存在着争议[20-22]。但目前广泛接受的观点为Fe5C2是费-托反应活性中心[22-25]。从表1可知,在Fe20/AlPO4、Fe20/γ-Al2O3和Fe20/MgAl2O4催化剂上CO转化率分别为2.85%、3.04%和5.36%,随着载体碱性增强CO转化率和STY上升,同时CH4的选择性依次降低,C5+的选择性和C2~C4的烯烷比(O/P)逐步升高。而3种催化剂的C2~C4烯烃选择性受到C5+的选择性和C2~C4的烯烷比的综合影响。
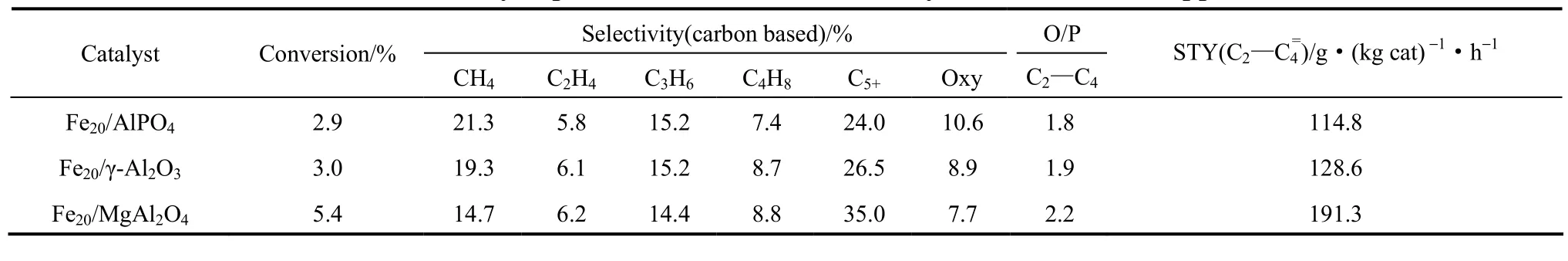
表1 不同负载型Fe基催化剂的催化反应性能结果Table 1 Catalytic performance of Fe-based catalysts with different supports
在费-托反应中常用ASF模型预测反应产物分布,使用碳链增长概率(α值)描述反应的碳增长过程。一般认为,甲烷由CO解离生成的C*物种加氢生成,甲醇由非解离的CO直接加氢生成,故甲烷等不经过链增长步骤的C1产物并未纳入链增长因子的拟合中。由图3可知,Fe20/AlPO4、Fe20/γ-Al2O3和Fe20/MgAl2O4的α值分别为0.60、0.61和0.68,随着载体碱性的增强α值不断增加,产物分布向长链方向增加。
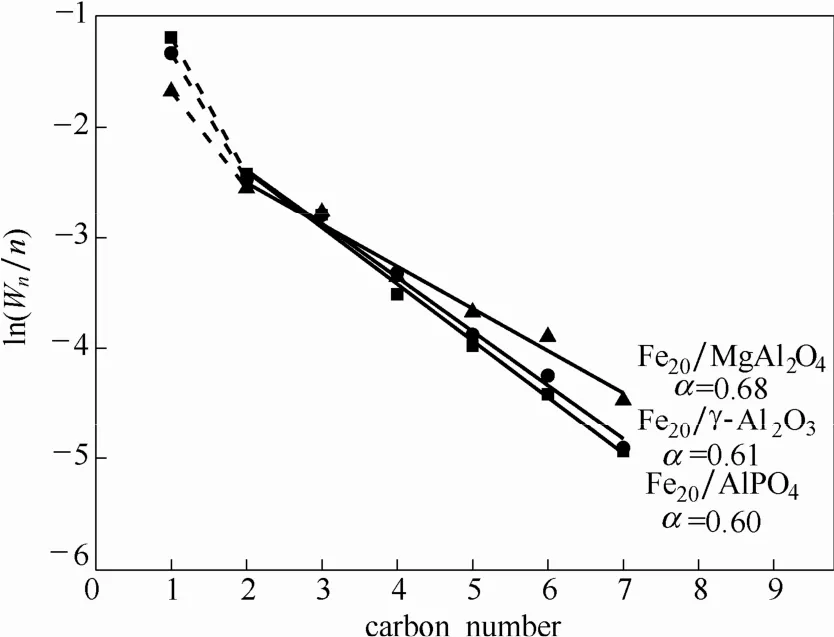
图3 负载型Fe基催化剂的ASF分布Fig.3 ASF distribution for supported Fe-based catalysts
2.3催化剂活性金属与载体的相互作用
2.3.1负载型Fe基催化剂的XRD表征图4为3种催化剂的XRD谱图。受到Fe负载量低和金属晶体大小的综合影响,XRD谱图中主要为载体的特征峰。在2θ为35.439°、62.545°处出现Fe3O4(01-079-0419)的特征衍射峰,可归属为Fe3O4的(311)、(440)晶面。催化剂在钝化过程中催化剂颗粒表面形成Fe2O3,但由于含量较低且并未影响到催化剂的体相结构,在XRD谱图中无法明显观察到长程有序的Fe2O3峰形。
2.3.2催化剂还原性能的比较图5为3种催化剂的H2-TPR谱图。由H2-TPR数据可见,由于不同载体与铁的相互作用强度不同,其还原性能也有所差异。图中第1个峰(700 K附近)归属为Fe2O3还原为Fe3O4,第2个峰(1000 K附近)归属为Fe3O4还原为α-Fe。AlPO4作为较强的酸性载体,其吸电子能力较强,内部电负性较大的PO4四面体结构与Fe作用,使得载体与活性金属之间的相互作用加强,即提升了铁氧化物的还原温度[26]。MgAl2O4由于Mg的引入比γ-Al2O3表现出更强的碱性,同时Mg、Al间又形成了特殊的尖晶石结构,这使得部分Fe物质进入尖晶石骨架中,增强了Fe物质的稳定性,强化了活性金属与载体的相互作用,降低了催化剂上铁氧化物的还原性[27]。因此Fe20/AlPO4和Fe20/MgAl2O4两种催化剂H2-TPR第2个峰的极值点未在实验测试温度范围内出现。
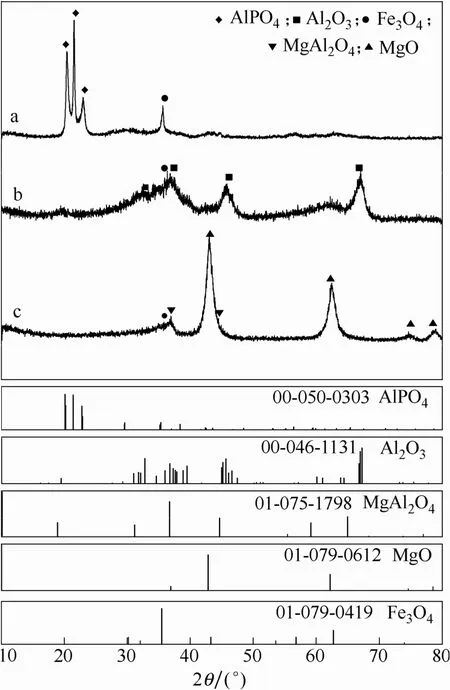
图4 Fe20/AlPO4(a)、Fe20/γ-Al2O3(b)和Fe20/MgAl2O4(c)催化剂的X射线衍射谱图Fig.4 XRD patterns of Fe20/AlPO4(a), Fe20/γ-Al2O3(b), and Fe20/MgAl2O4(c)

图5 负载型Fe基催化剂H2程序升温还原谱图Fig.5 H2-TPR profiles of supported Fe-based catalysts
2.3.3催化剂的碱性分析图6为3种负载型Fe基催化剂的CO2-TPD谱图。图中第1个峰(390 K附近)归属为载体与CO2之间的弱吸附,第2、3个峰(800 K和927 K附近)归属为活性金属上的CO2强吸附。对比图2可知,由于Fe及相应碳物质的加入覆盖了一定量的载体碱性位,使得载体的CO2脱附峰温度降低,但催化剂表面的碱性环境依然与载体碱性相对应。
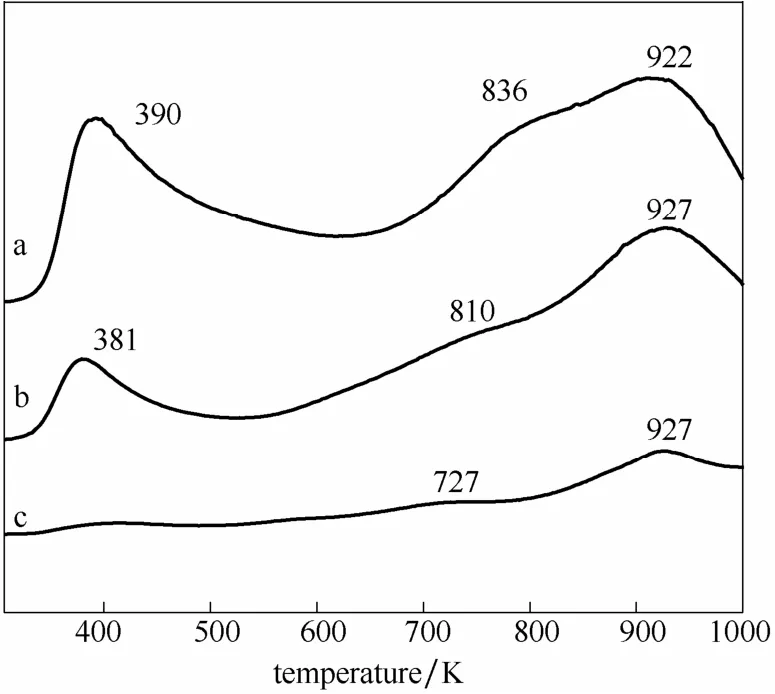
图6 Fe20/MgAl2O4(a)、Fe20/γ-Al2O3(b)和Fe20/AlPO4(c)的CO2-TPD谱图Fig.6 CO2-TPD profiles of Fe20/MgAl2O4(a), Fe20/γ-Al2O3(b), and Fe20/AlPO4(c)
2.3.4新鲜催化剂上碳层的分析制备过程中前体C6H11FeNO7的使用会使高温煅烧后的催化剂依然存在有碳层。从XRD谱图分析可知催化剂体相中没有大量的成晶型的碳物种。本工作使用Raman光谱技术对催化剂中表面碳层进行分析。图7为3种催化剂的Raman谱图。通常1364 cm−1和1597 cm−1附近的拉曼振动峰同时出现,代表碳材料中的D峰和G峰。2089 cm−1归属为连续的双键烃链[28]。从Raman谱图分析可知,新鲜催化剂表层既具有石墨化碳又具有无定形碳。随着载体碱性的增强,ID/IG有小幅度的降低,分别为0.6025、0.5820、0.5763。依据Tuinstra & Koenig关系[29],表明催化剂表层有序石墨片层平面相关长度Ld的增加。G 峰的半峰宽也随载体碱性增强而轻微减小,表明催化剂上碳的无序化程度增加。而且,载体碱性越弱,碳sp键的Raman峰强越弱。
上述结果说明载体的碱性即给电子能力不同使得柠檬酸铁铵在高温煅烧过程中形成碳层的厚度及石墨化程度不同。这也间接反映了在不同给电子能力的载体上Fe的配位环境不同,导致了Fe与C之间相互作用的差异。
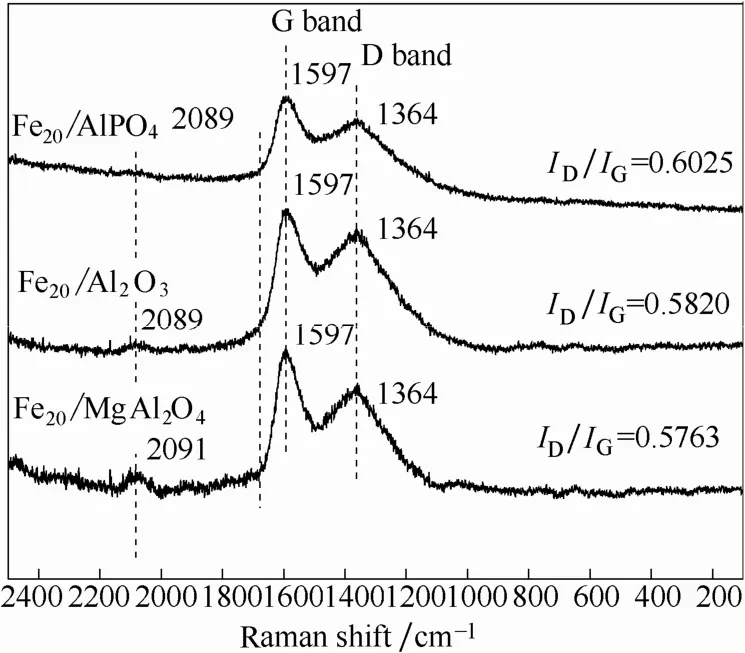
图7 新鲜催化剂的Raman谱图Fig.7 Raman spectra of fresh catalysts

图8 Fe20/AlPO4(a)、Fe20/γ-Al2O3(b)和Fe20/MgAl2O4(c)催化剂的TPH谱图Fig.8 TPH profiles of Fe20/AlPO4(a), Fe20/γ-Al2O3(b) and Fe20/MgAl2O4(c)
通过TPH谱图半定量分析可知,随着催化剂载体碱性的增强,表面活性碳物质(吸附碳、原子碳)的含量降低,惰性碳物质(石墨碳)的含量增加。
载体碱性的不同会导致对活性金属电子效应的差异。表面碱性的增强可能提高Fe相的电子密度,强化Fe—C键,进而削弱C—O键,因而提高了CO在催化剂表面的吸附解离能力(削弱了H2在表面的吸附解离),提高了反应中间产物碳链增长概率[31]。这一效应表现为C5+选择性随载体碱性提高而增加。
此外,载体对活性金属的给电子能力差异导致在活性金属上不同碳物质含量的差异。载体碱性增强,吸附碳的数量减少。基于Mars-van Krevelen模
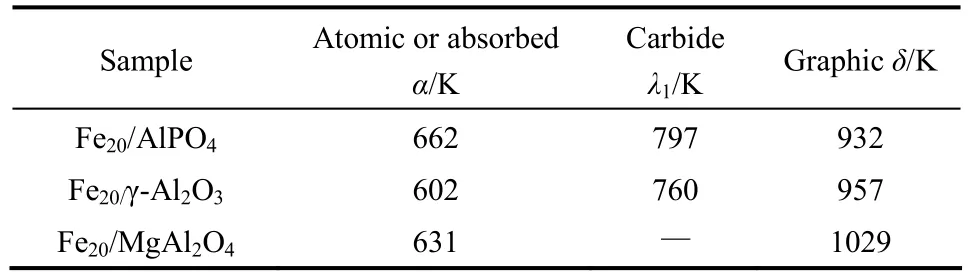
表2 不同新鲜Fe基催化剂的程序升温加氢结果Table 2 TPH results of fresh Fe-based catalysts
2.4负载性Fe基催化剂构效关系的建立
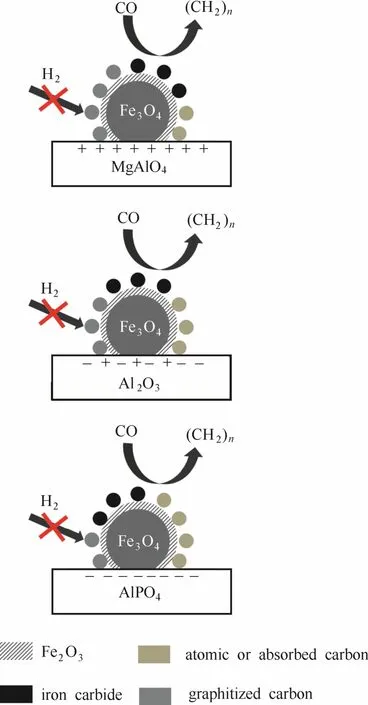
图9 负载型Fe基催化剂构效关系Fig. 9 Scheme of structure-performance relationship of catalysts
载体的碱性对Fe基费-托催化剂的催化性能具有重要影响。依据本工作制备而得的新鲜催化剂是以Fe3O4颗粒为主体,表面覆盖有Fe2O3和碳薄膜的负载型功能性材料。催化剂在623 K 条件下还原5 h后,催化剂中的Fe2O3的含量进一步减少,同时催化剂表面的吸附碳及原子碳含量也随碳物质加氢而减少。载体对催化剂结构及性能影响如图9所示。型[32],Ordomsky等[17]认为费-托合成反应中的链引发与铁碳化合物中的碳有关。Xu等[30]研究显示,沉积碳的增加、吸附碳的减少有利于形成铁碳化合物,从而提高催化剂的CO转化率。同时,载体碱性增强也使得催化剂表面碳物质石墨化程度增加,石墨层占据了一定量的二次吸附位,抑制了烯烃的二次加氢反应,使得费-托反应产物中的烯烷比增加。
3 结 论
(1)AlPO4、γ-Al2O3、MgAl2O4为碱性逐渐增强的3种载体,依托3种载体制备了负载型Fe 基费-托催化剂,并进行了催化性能评价。实验结果表明,随着载体碱性的增强,催化剂活性增强,反应产物向长链方向增长,烯烷比增加,提高C2~C4烯烃选择性需要同时控制碳链增长和增加烯烷比。
(2)以柠檬酸铁铵为前体的催化剂经过煅烧后表面会有残留碳,通过对碳物种分析可知新鲜催化剂表面上含有吸附碳(原子碳)、碳化铁、石墨碳3种碳物质。载体的碱性越强,煅烧后催化剂表层碳物种中吸附(原子)碳数量越少,石墨化程度越深。
(3)建立了基于不同酸碱性载体的催化剂构效关系。载体的给电子能力影响活性金属的催化性能。高表面碱性将增强CO在催化剂表面的吸附解离能力,进而提高C5+选择性,而因载体碱性引起催化剂表面碳物质含量差异,从而进一步影响烯烃再吸附能力及二次加氢能力。
一些旅游开发者对旅游地设施没有投入足够的资金,没有建立旅游地应该配置的设施[2],如垃圾池、排水设施、旅游步道等,旅游地呈现出杂乱无章的景象。这些情况制约了旅游业健康发展。
符号说明
Ld——有序石墨片层平面相关长度
Mi——出口气体中各低碳烯烃的摩尔质量,g·mol−1
mcat——催化剂装填量,kg
n ——物质i的碳数
Qin(out)——反应器进(出)口气流量,ml·h−1
SBET——BET比表面积,m2·g−1
Si——物质i的选择性,%
STY ——产物的时空收率,g·(kg cat)−1·h−1
Vtotal——总孔容,cm3·g−1
vCO——原料气中CO的体积分数
vi——物质i在出口产物中的体积分数
vo,i——出口气体中各低碳烯烃的体积分数
Wn——产物的质量分数,%
XCO——CO转化率,%
α ——链增长概率
References
[1] COMA A, MELO F V, SAUVANAUD L, et al. Light cracked naphtha processing: controlling chemistry for maximum propylene production [J]. Catalysis Today, 2005, 107-108: 699-706.
[2] DIERCKS R, ARNDT J D, FREYER S, et al. Raw material changes in the chemical industry [J]. Chemical Engineering & Technology, 2008, 31(5): 631-637.
[3] WANG S, ZHU Z H. Catalytic conversion of alkanes to olefins by carbon dioxide oxidative dehydrogenation: a review [J]. Energy & Fuels, 2004, 18(4): 1126-1139.
[4] 金涌, 周禹成, 胡山鹰. 低碳理念指导的煤化工产业发展探讨 [J].化工学报, 2012, 63(1): 3-8. DOI: 10.3969/j.issn. 0438-1157. 2012.01.001. JIN Y, ZHOU Y C, HU S Y. Discussion on development of coal chemical industry using low-carbon concept [J]. CIESC Journal, 2012, 63(1): 3-8. DOI: 10.3969/j.issn.0438-1157.2012.01.001.
[5] KUNKES E L, SIMONETTI D A, WEST R M, et al. Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes [J]. Science, 2008, 322(5900): 417-421.
[6] LI P, ZHANG W, HAN X, et al. Conversion of methanol to hydrocarbons over phosphorus-modified ZSM-5/ZSM-11 intergrowth zeolites [J]. Catalysis Letters, 2010, 134(1/2): 124-130.
[7] DUPAIN X, KRUL R A, SCHAVERIEN C J, et al. Production of clean transportation fuels and lower olefins from Fischer-Tropsch synthesis waxes under fluid catalytic cracking conditions: the potential of highly paraffinic feedstocks for FCC [J]. Applied Catalysis B: Environmental, 2006, 63(3): 277-295.
[8] GALVIS H M T, BITTER J H, Khare C B, et al. Supported iron nanoparticles as catalysts for sustainable production of lower olefins [J]. Science, 2012, 335(6070): 835-838.
[9] TORRES GALVIS H M, DE JONG K P. Catalysts for production of lower olefins from synthesis gas: a review [J]. ACS Catalysis, 2013, 3(9): 2130-2149.
[10] SUN B, YU G, LIN J, et al. A highly selective Raney Fe@ HZSM-5 Fischer–Tropsch synthesis catalyst for gasoline production: one-pot synthesis and unexpected effect of zeolites [J]. Catalysis Science & Technology, 2012, 2(8): 1625-1629.
[11] BARRAULT J, FORQUY C, MENEZO J C, et al. Hydrocondensation of CO2(CO) over supported iron catalysts [J]. Reaction Kinetics and Catalysis Letters, 1981, 17(3/4): 373-378.
[12] BUKUR D B, LANG X, MUKESH D, et al. Binder/support effects on the activity and selectivity of iron catalysts in the Fischer-Tropsch synthesis [J]. Industrial & Engineering Chemistry Research, 1990, 29(8): 1588-1599.
[13] CUBEIRO M L, LóPEZ C M, COLMENARES A, et al. Use of aluminophosphate molecular sieves in CO hydrogenation [J]. Applied Catalysis A: General, 1998, 167(2): 183-193.
[14] SOMMEN A P B, STOOP F, VAN DER WIELE K. Synthesis gas conversion on carbon supported iron catalysts and the nature of deactivation [J]. Applied Catalysis, 1985, 14: 277-288.
[15] SUN P, SIDDIQI G, CHI M, et al. Synthesis and characterization of a new catalyst Pt/Mg(Ga)(Al)O for alkane dehydrogenation [J]. Journal of Catalysis, 2010, 274(2): 192-199.
[16] MACHIDA M, MINAMI S, HINOKUMA S, et al. Unusual redox behavior of Rh/AlPO4and its impact on three-way catalysis [J].Journal of Physical Chemistry C, 2014, 119(1): 373-380.
[17] ORDOMSKY V V, LEGRAS B, CHENG K, et al. The role of carbon atoms of supported iron carbides in Fischer-Tropsch synthesis [J]. Catalysis Science & Technology, 2015, 5(3): 1433-1437.
[18] HERINGTON E F G. The Fischer-Tropsch synthesis considered as a polymerization reaction [J]. Chemistry & Industry, 1946, 65: 346-347.
[19] ANDERSON R B, FRIEDEL R A, STORCH H H. Fischer-Tropsch reaction mechanism involving stepwise growth of carbon chain [J]. Journal of Chemical Physics, 1951, 19(3): 313-319.
[20] DE S E. The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour [J]. Chemical Society Reviews, 2008, 37(12):2758-2781.
[21] KUIVILA C S, STAIR P C, BUTT J B. Compositional aspects of iron Fischer-Tropsch catalysts: an XPS reaction study [J]. Journal of Catalysis, 1989, 118(2): 299-311.
[22] XU K, SUN B, LIN J, et al. ε-Iron carbide as a low-temperature Fischer-Tropsch synthesis catalyst [J]. Nature Communications, 2014, 5:5783-5783.
[23] SHROFF M D , KALAKKAD D S, COULTER K E, et al. Activation of precipitated iron Fischer-Tropsch synthesis catalysts [J]. Journal of Catalysis, 1995, 156(2):185–207.
[24] YANG C, ZHAO H, HOU Y, et al. Fe5C2nanoparticles: a facile bromide-induced synthesis and as an active phase for Fischer-Tropsch synthesis [J]. Journal of the American Chemical Society, 2012, 134(38): 15814-15821.
[25] SUN B, LIN J, XU K, et al. Fischer–Tropsch synthesis over skeletal FeCe catalysts leached from rapidly quenched ternary Fe Ce Al alloys [J]. ChemCatChem, 2013, 5(12): 3857-3865.
[26] JI M, CHEN G, WANG J, et al. Dehydrogenation of ethylbenzene to styrene with CO2over iron oxide-based catalysts [J]. Catalysis Today, 2010, 158(3): 464-469.
[27] 陈桂丽, 陈鑫, 王新葵,等. 铁系催化剂上乙苯与CO2氧化脱氢反应 [J]. 催化学报, 2009, 30(7):619-623. DOI:10.3321/j.issn:0253-9837.2009.07.007. CHEN G L, CHEN X, WANG X K, et al. Oxidative dehydrogenation of ethylbenzene with CO2over iron oxide-based catalysts [J]. Chinese Journal of Catalysis, 2009, 30(7):619-623. DOI:10.3321/j.issn: 0253-9837.2009.07.007.
[28] RAVAGNAN L, SIVIERO F, LENARDI C, et al. Cluster-beam deposition and in situ characterization of carbyne-rich carbon films [J]. Physical Review Letters, 2002, 89(28):287-291.
[29] TUINSTRA F, KOENIG J L. Raman spectrum of graphite [J]. Journal of Chemical Physics, 1970, 53(3):1126-1130.
[30] XU J, BARTHOLOMEW C H. Temperature-programmed hydrogenation (TPH) and in situ Mössbauer spectroscopy studies of carbonaceous species on silica-supported iron Fischer-Tropsch catalysts [J]. Journal of Physical Chemistry B, 2005, 109(6): 2392-2403.
[31] LU J, YANG L, XU B, et al. Promotion effects of nitrogen doping into carbon nanotubes on supported iron Fischer–Tropsch catalysts for lower olefins [J]. ACS Catalysis, 2014, 4(2): 613-621.
[32] GRACIA J M, PRINSLOO F F, Niemantsverdriet J W. Mars-van Krevelen-like mechanism of CO hydrogenation on an iron carbide surface [J]. Catalysis Letters, 2009, 133(3/4): 257-261.
DOI:10.11949/j.issn.0438-1157.20151251
中图分类号:TQ 018
文献标志码:A
文章编号:0438—1157(2016)02—0549—08
基金项目:国家自然科学基金项目(21273070,21576084)。
Corresponding author:Prof. HAN Yifan, yifanhan@ecust.edu.cn supported by the National Natural Science Foundation of China (21273070, 21576084).
Effect of support basicity on iron-based catalysts for Fischer-Tropsch synthesis
ZHANG Jun, ZHANG Zhengpai, SU Junjie, FU Donglong, DAI Weiwei, LIU Da, XU Jing, HAN Yifan
(State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China)
Abstract:Lower hydrocarbons are key building blocks in chemical industry. The Fischer-Tropsch synthesis has been considered as one of the most promising non-oil based routes for lower hydrocarbons production. Previous studies have demonstrated that the supports can greatly affect the product distribution. In this work, the effects of the base property of supports on the catalytic performance of different Fe-based catalysts (Fe20/AlPO4, Fe20/γ-Al2O3and Fe20/MgAl2O4) were investigated. The results showed that, with the increase in support basicity, the chain growth probability (α value), the selectivity of C5+hydrocarbons and the olefins/paraffin ratio increased. Moreover, by combining with the Raman and temperature programmed hydrogenation (TPH), it was found that the higher basicity of supports could lead to the less active carbon (absorptive and atomic carbon) and the more inactive carbon (graphitized carbon) formation. In addition, with the combination of other characterization, such as XRD, H2-TPR, CO2-TPR, the structure-performance relationship was constructed. The variation in electron-donating ability of the supports strongly affected the content of carbonaceous species on the catalyst surface and the metal-support interaction, thereby leading to the difference in catalytic activity and selectivity.
Key words:Fischer-Tropsch synthesis; syngas; catalysis; Fe-based catalysts; catalyst support; basicity; carbon specie
猜你喜欢
华人时刊(2022年9期)2022-09-06
华人时刊(2020年15期)2020-12-14
工业水处理(2020年4期)2020-03-09
石油石化绿色低碳(2019年6期)2019-02-13
中国有色金属学报(2018年2期)2018-03-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
浙江大学学报(工学版)(2016年11期)2016-06-05
浙江农业科学(2016年11期)2016-05-04
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
