
TRPC6基因突变致局灶节段性肾小球硬化的研究进展
2016-06-23 13:25李贵森
实用医院临床杂志 2016年3期
彭 梅,李贵森
(1.遵义医学院,贵州 遵义 563000;2.四川省医学科学院·四川省人民医院肾脏内科暨肾脏病研究所,四川 成都 610072)
TRPC6基因突变致局灶节段性肾小球硬化的研究进展
彭 梅1,2,李贵森2△
(1.遵义医学院,贵州 遵义 563000;2.四川省医学科学院·四川省人民医院肾脏内科暨肾脏病研究所,四川 成都 610072)
局灶节段性肾小球硬化(focal segmental glomerulosclerosis,FSGS)是激素耐药型肾病综合征的主要病因,发病机制十分复杂。关键的足细胞结构蛋白基因突变常常导致遗传性FSGS,如裂孔隔膜(SD)的瞬时受体电位阳离子通道蛋白6(TRPC6)即为其中之一。TRPC6基因突变可以导致细胞钙内流增加促使FSGS 的发生。现就TRPC6基因突变致FSGS发病机制的研究进展作一综述。
局灶节段性肾小球硬化;TRPC6;突变;发病机制
局灶节段性肾小球硬化(focal segmental glomerulosclerosis,FSGS)是肾病综合征常见的原因之一,也是导致激素抵抗型肾病综合征主要病因之一,预后较差,可迅速进展为终末期肾脏病(end-stage renal disease,ESRD)[1~3]。近年来FSGS发病呈上升趋势,占我国肾活检中原发性肾小球疾病的比例为6%~12%,是儿童及中青年ESRD的主要原因之一[2,4,5]。FSGS是以局灶节段分布的肾小球硬化为基本病理改变的一组肾小球病变[6,7],病因包括原发性、继发性和遗传性。遗传性FSGS也叫家族性FSGS。目前认为,不论是家族性FSGS,还是散发性FSGS,遗传因素在其发病中可能都起重要作用,研究发现FSGS可以表现为明显的家族聚集性和种族差异,例如非洲裔美国人的发病率明显高于高加索人[8~10]。1992年Mathis等[11]首先报道FSGS可以表现为常染色体显性遗传模式。Brown等[12]在此基础上克隆出第一个FSGS的致病基因ACTN4,其编码的蛋白为α辅肌动蛋白4(α-actinin 4)。此后越来越多的FSGS 致病基因被克隆,迄今已发现数十余种基因突变参与FSGS的发病,包括TRPC6,INF2等[8,10,13~16]。这些基因突变的深入研究,对进一步阐明FSGS的发病机制具有重要作用。Winn等[17]首先从家族性FSGS患者中克隆出了一个导致家族性局灶节段性肾小球硬化(FSGS)的新基因--TRPC6,其编码蛋白瞬时受体电位阳离子通道蛋白(transient receptor potential cation channel 6,TRPC6)是一个非选择性阳离子通道,在足细胞表达,与裂孔隔膜分子Nephrin以及Podocin间存在相互作用,共同参与足细胞间的信号传导、细胞极化和稳定骨架结构等生理功能[18]。因此,认识该基因、筛查可能存在的突变以及进一步研究该基因的功能将对探讨FSGS的发病机制以及防治具有重要意义。本文着重介绍TRPC6致FSGS基因突变种类研究,为进一步探究其致FSGS的发病机制及防治提供理论基础。
1 TRPC6相关基因突变研究
1.1 基本概述 瞬时受体电位(transient receptor potential,TRP)是一种新发现的细胞膜上的钙通道,在各种生物细胞中有着广泛的表达。哺乳动物TRP通道蛋白包括6个关联的蛋白家族:TRPC、TRPV、TRPM、TRPP、TRPN以及TRPML,其中每个家族又有许多亚型。TRPC是TRP家族的主要成员之一,它的激活机制依赖于磷脂酶C(phospholipase C,PLC)信号通路,被认为是最可能的钙库操纵性钙通道和受体操纵性钙通道的分子基础。钙离子是细胞内的第二信使,广泛存在于从细菌到特殊神经元等各类细胞中,在哺乳动物机体内有广泛表达[19]。TRPC包括7个亚家族,即TRPC1~7,其中TRPC3和TRPC6在TRPC亚家族中比较有代表性[20~22],也是目前国际研究中较受关注的2个亚型。人TRPC6 定位于染色体11q21~q22,共132365个碱基(GeneBank:NG_011476),含13个外显子[23]。TRPC6的编码蛋白是一个含有931个氨基酸的通道蛋白,为6次跨膜蛋白,其N端和C端均在胞内,由第5和第6跨膜结构域共同构成非选择性阳离子通道[24]。
1.2 突变种类 近年的研究陆续报道一些新的TRPC6突变与FSGS发病相关以来,迄今为止,已经证实15种TRPC6 基因突变参与FSGS发病(图1)。其中,3个突变(G109S、P112Q、N125S)位于第1个锚蛋白重复区域(ankyrin repeat,ANK)区域,2个突变(M132T、N143S)位于ANK2区域,1个突变(R175Q)在ANK3区域,1个突变(H218L)位于ANK4区域,另外2个突变(S270T、L395A)位于细胞质N-末端[17,25~29];余下6个突变(L780P、K874X、Q889K、R895C、R895L、E897K)位于细胞质C-末端[17,26,27,30]。

图1 FSGS相关的TRPC6基因突变
2 TRPC6突变致FSGS发病的机制
2.1 TRPC6突变影响钙离子信号通路传导 2005年Winn等[17]首先发现P112Q突变细胞内钙离子浓度、钙离子峰浓度、细胞电流及TRPC6蛋白表达较突变型较野生型明显升高。目前,通过电生理现象和/或细胞内Ca2+测量,上述15种TRPC6基因突变中的10个突变(P112Q、N143S、R895C、E897K、Q889K、M132T、N125S、H218L、R895L、R175Q突变)均可以导致钙通道活性增加。因此,TRPC6通道是最可能的钙库操纵性钙通道和受体操纵性钙通道。TRPC6基因突变可以影响TRPC6的Ca2+通道活性,导致Ca2+内流增加,从而激活Ca2+依赖信号通路,最终导致FSGS的发生。
TRP通道与多种生物功能有关,如细胞生长、离子平衡、机械感受和依赖PLC的钙内流等[17];钙作为第二信使可以影响细胞内上述的大多数功能[17,31]。血管紧张素II通过AT1受体,在包括FSGS在内的一些肾脏疾病的损伤过程中(如蛋白尿的产生及肾脏损伤的进展)起着关键作用[17,32]。AT1受体与异源三聚体鸟嘌呤核苷酸-G蛋白结合,激活磷脂酶C-β(PLC-β),水解磷脂酰肌醇异构体4,5-二磷酸磷脂酰肌醇(PIP2),产生三磷酸肌醇(InsP3)和二酰甘油(DAG)。InsP3可迅速地从细胞膜内侧向胞质溶胶中扩散,一方面打开细胞膜上的钙通道使Ca2+进入细胞内,另一方面开启细胞内钙池(内质网)增加Ca2+的释放;同时DAG也促进细胞内钙池释放,增加细胞内Ca2+,即InsP3和DAG两者协同提高胞内游离钙的浓度。胞质Ca2+含量的上升,激活一种称为钙调蛋白的Ca2+结合蛋白,后者可调节其他酶类的活性,并最终导致钙调磷酸酶(CaN)的激活[17,33]。Santin等[26]在儿童和成人的非家族性FSGS中究发现的G109S、N125S、L780P突变均为错义突变。,研究发现有此3种基因突变的患者对钙调磷酸酶抑制剂(CNI)有较好的疗效也进一步证明TRPC6突变可能使钙信号增加,激活CaN,促进细胞凋亡,破坏了足细胞肌动蛋白细胞骨架[34],同时阻断CaN活性可能改善蛋白尿,这与临床上使用环孢素、他克莫司等钙调磷酸酶抑制剂后部分TRPC6基因突变的FSGS患者蛋白尿获得明显缓解的一致性进一步证实上述可能。由此可见,TRPC6突变在钙信号通路传导中发挥非常重要的作用。
2.2 突变影响CaN-NFAT信号通路 活化细胞核转录因子(nuclear factor of activated T cells,NFAT)是一类重要的转录因子,广泛分布于机体的免疫及非免疫细胞,其活性受钙离子依赖性的CaN的调节,主要通过受体-Ca-CaN信号途径被激活,从而发生其基因调控功能。Koitabashi等[35]研究发现二脂酰甘油(DAG)激活TRPC6通道上的Gq蛋白偶联受体,导致Ca2+进入钙池,钙离子内流可激活丝氨酸-苏氨酸-CaN,这可以使NFAT家族的转录因子去磷酸化,去磷酸化NFAT入核,进一步激活某一基因程序,提供正反馈回路,进一步刺激TRPC6电流和相关的信号。另外,Schlöndorff等[36]提出TRPC6突变相关的FSGS是由于CaN-NFAT信号途径的激活并证实了以往报道的P112Q、R895C和E897K突变能够显着地刺激NFAT活化,说明FSGS TRPC6突变的确增强NFAT活化;该研究还发现环孢霉素A及FK506抑制TRPC6介导的NFAT活化,这与Gigante等[27]报道的临床表现为SRNS的H218L突变患儿,对钙调磷酸酶抑制剂治疗敏感表现一致。以上信息说明CaN-NFAT信号通路在调节肾小球功能方面具有非常重要的作用。也为这些FSGS患者发病机理及未来的治疗提供重要的线索。
2.3 突变致足细胞结构功能改变 已经报道TRPC6 mRNA在多种组织中表达,包括肾脏、脑、肺、卵巢等[17,37~39]。Winn等用针对人TRPC6的抗体证实TRPC6在肾小球和肾小管均有表达,进一步的荧光原位杂交(fluorescence in situ hybridization,FISH)实验表明TRPC6 mRNA在肾小球和肾小管弥漫性表达,与免疫荧光染色几乎相同。Reiser等[25]应用免疫荧光双标记显示在足细胞TRPC6与Nephrin、Podocin和CD2AP存在共定位分布,免疫沉淀结果还提示TRPC6与Nephrin、Podocin有相互作用,而与CD2AP间无相互作用。此外,他们还发现Nephrin敲除小鼠足细胞内TRPC6表达增加而且分布也发生了改变,呈颗粒聚集样表现,这提示Nephrin缺失诱发了TRPC6表达的增加和分布的改变。另有研究[27]发现H218L、R895L突变型患者肾小球的TRPC6表达明显上调,nephrin表达却明显下调,同时观察到nephrin的蛋白质沿毛细血管袢不规则分布;其中表现为R895L突变的塌陷型患儿的TRPC6在新月形上皮细胞明显上调。以上事实表明TRPC6属于裂孔隔膜蛋白,它与Nephrin、Podocin等重要的裂孔隔膜分子共同组成一个信号传导复合物以维持足突和裂孔隔膜结构和功能的完整性。TRPC6突变导致足细胞钙内流而引起细胞骨架过度收缩,使得细胞骨架对肾小球滤过静水压作出失适应性反应,最终引起足细胞结构和功能受损;TRPC6突变蛋白与Nephrin、Podocin等裂孔隔膜分子相互作用发生紊乱,从而干扰了由它们所组成的裂孔隔膜信号复合体的功能,最终导致足细胞不能维持正常的生物学功能。
3 TRPC6突变所致FSGS临床特征分析
TRPC6基因突变在FSGS患者中的发生情况还不太清楚,不同研究报道的差异较明显。我们总结了目前所报道的家族性FSGS和散发性FSGS患者中TRPC6突变的情况,见表1。根据家族史分析,所有家系的遗传方式均为常染色体显性遗传,每个氨基酸的取代均为进化上的高度保守的残基。存在携带有TRPC6突变但无临床表现的个体。中国TRPC6突变占家族性FSGS的比例约3.2%,其他种族约为7%。这些家族患病个体在30~40岁开始出现大量蛋白尿,其中大约60%进展为ESRD,从开始出现临床表现至进展为ESRD时间约为10年。患者肾脏病理表现多为FSGS。散发性FSGS患者无特异性表现。M132T突变是首次报道的可以导致早发型的FSGS,早发型FSGS多表现为早发(最小发病年龄为1岁)和SRNS,但对钙调磷酸酶抑制剂(如环孢素A、FK506)治疗敏感;其中早发型FSGS肾脏活检为塌陷型的肾小球硬化患者可迅速进展为ESRD。
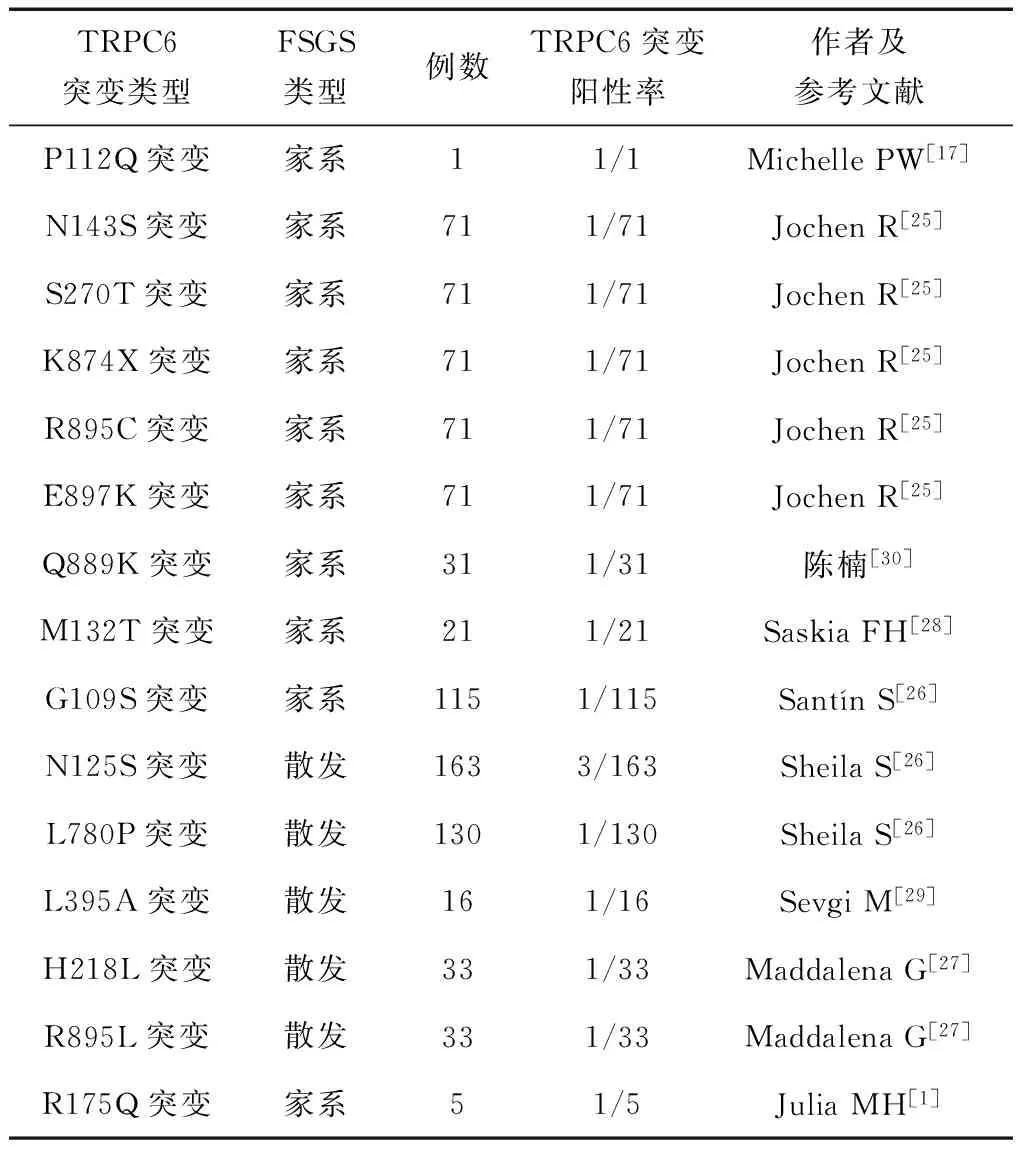
表1 TRPC6基因突变致FSGS临床总结
注:家系研究的例数是指研究所纳入的家系数;家系中的突变阳性率指所有家系中发生突变的家系数。
目前有关TRPC6基因突变的研究取得较大进展,但是TRPC6基因突变与FSGS的确切发生机制仍不十分清楚,需要更多的体外功能试验甚至建立动物模型试验来明确,同时FSGS的进一步防治也需要进一步探索。
[1] Hofstra JM.New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis[J].Nephrol Dial Transplant,2013,28(7):1830-1838.
[2] D′Agati VD,Kaskel FJ,Falk RJ.Focal segmental glomerulosclerosis[J].N Engl J Med,2011,365(25):2398-2411.
[3] Haas M.Changing etiologies of unexplained adult nephrotic syndrome:a comparison of renal biopsy findings from 1976-1979 and 1995-1997[J].Am J Kidney Dis,1997,30(5):621-631.
[4] Zhou FD.The changing spectrum of primary glomerular diseases within 15 years:a survey of 3331 patients in a single Chinese centre[J].Nephrol Dial Transplant,2009,24(3):870-876.
[5] 陈惠萍.10594例肾活检病理资料分析[J].肾脏病与透析肾移植杂志,2000,9(6):501-509.
[6] D′Agati VD.Pathologic classification of focal segmental glomerulosclerosis:a working proposal[J].Am J Kidney Dis,2004,43(2):368-382.
[7] Deegens JK.Pathological variants of focal segmental glomerulosclerosis in an adult Dutch population-epidemiology and outcome[J].Nephrol Dial Transplant,2008,23(1):186-192.
[8] Pollak MR.Focal segmental glomerulosclerosis:recent advances[J].Curr Opin Nephrol Hypertens,2008,17(2):138-142.
[9] Haas M.Changing etiologies of unexplained adult nephrotic syndrome:a comparison of renal biopsy findings from 1976-1979 and 1995-1997[J].Am J Kidney Dis,1997,30(5):621-631.
[10]李贵森,王庆文.局灶节段性肾小球硬化的遗传背景[J].肾脏病与透析肾移植杂志,2010,19(4):373-377.
[11]Mathis BJ,Calabrese KE,Slick GL.Familial glomerular disease with asymptomatic proteinuria and nephrotic syndrome:a new clinical entity[J].J Am Osteopath Assoc,1992,92(7):875-884.
[12]Kaplan JM.Mutations in ACTN4,encoding alpha-actinin-4,cause familial focal segmental glomerulosclerosis[J].Nat Genet,2000,24(3):251-256.
[13]Brown EJ.Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis[J].Nat Genet,2010,42(1):72-76.
[14]Lowik MM.Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis-a review[J].Eur J Pediatr,2009,168(11):1291-1304.
[15]Kopp JB.MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis[J].Nat Genet,2008,40(10):1175-1184.
[16]Woroniecki RP,Kopp JB.Genetics of focal segmental glomerulosclerosis[J].Pediatr Nephrol,2007,22(5):638-644.
[17]Winn MP.A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis[J].Science,2005,308(5729):1801-1804.
[18]Huber TB,Schermer B,Benzing T.Podocin organizes ion channel-lipid supercomplexes:implications for mechanosensation at the slit diaphragm[J].Nephron Exp Nephrol,2007,106(2):e27-31.
[19]张瑶,于力.瞬时受体电位阳离子通道蛋白6与肾脏疾病[J].临床儿科杂志,2010,28(6):594-597.
[20]Moller CC.Induction of TRPC6 channel in acquired forms of proteinuric kidney disease[J].J Am Soc Nephrol,2007,18(1):29-36.
[21]Kim YH.Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat[J].Kidney Int,2001,60(3):957-968.
[22]Pagtalunan ME.Podocyte loss and progressive glomerular injury in type II diabetes[J].J Clin Invest,1997,99(2):342-348.
[23]Beech DJ.Emerging functions of 10 types of TRP cationic channel in vascular smooth muscle[J].Clin Exp Pharmacol Physiol,2005,32(8):597-603.
[24]Estacion M.Activation of human TRPC6 channels by receptor stimulation[J].J Biol Chem,2004,279(21):22047-22056.
[25]Reiser J.TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function[J].Nat Genet,2005,37(7):739-744.
[26]Santin S.TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis[J].Nephrol Dial Transplant,2009,24(10):3089-3096.
[27]Gigante M.TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype[J].Clin J Am Soc Nephrol,2011,6(7):1626-1634.
[28]Heeringa SF.A novel TRPC6 mutation that causes childhood FSGS[J].PLoS One,2009,4(11):e7771.
[29]Mir S.TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome[J].Nephrol Dial Transplant,2012,27(1):205-209.
[30]Zhu B.Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients[J].Mutat Res,2009,664(1-2):84-90.
[31]Yamamoto M,Hara H,Adachi T.The expression of extracellular-superoxide dismutase is increased by lysophosphatidylcholine in human monocytic U937 cells[J].Atherosclerosis,2002,163(2):223-228.
[32]Taal MW,Brenner BM.Renoprotective benefits of RAS inhibition:from ACEI to angiotensin II antagonists[J].Kidney Int,2000,57(5):1803-1817.
[33]Balla T.Signaling events activated by angiotensin II receptors:what goes before and after the calcium signals[J].Endocr Res,1998,24(3-4):335-344.
[34]Faul C.The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A[J].Nat Med,2008,14(9):931-938.
[35]Koitabashi N.Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition[J].J Mol Cell Cardiol,2010,48(4):713-724.
[36]Schlondorff J.TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription[J].Am J Physiol Cell Physiol,2009,296(3):C558-569.
[37]Conlon PJ.Spectrum of disease in familial focal and segmental glomerulosclerosis[J].Kidney Int,1999,56(5):1863-1871.
[38]Riccio A.mRNA distribution analysis of human TRPC family in CNS and peripheral tissues[J].Brain Res Mol Brain Res,2002,109(1-2):95-104.
[39]Garcia RL,Schilling WP.Differential expression of mammalian TRP homologues across tissues and cell lines[J].Biochem Biophys Res Commun,1997,239(1):279-283.
Mutations of TRPC6 and pathogenesis of focal segmental glomerulosclerosis
PENG Mei1,2,LI Gui-sen2
四川省青年科技创新研究团队基金资助项目(编号:2015TD0013)
R692.6
B
1672-6170(2016)03-0170-04
2015-09-28;
2015-11-30)
△通讯作者
猜你喜欢
中国临床医学影像杂志(2021年10期)2021-11-22
中国临床医学影像杂志(2021年6期)2021-08-14
中国生殖健康(2020年2期)2021-01-18
现代临床医学(2019年4期)2019-09-10
国际呼吸杂志(2019年2期)2019-02-14
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中西医结合心血管病杂志(电子版)(2015年35期)2015-01-21
安徽医专学报(2014年6期)2014-03-20
河南医学研究(2014年5期)2014-02-27
