
5-硝基-3-三硝甲基-1H-1,2,4-三唑和5,5′-双(三硝甲基)-3,3′-偶氮-1H-1,2,4-三唑的合成与理论研究
2016-05-11 09:12姚二岗丁可伟苏海鹏李陶琦葛忠学
含能材料 2016年1期
肖 啸, 姚二岗,2, 刘 庆, 丁可伟, 苏海鹏, 李陶琦, 张 敏, 葛忠学
(1. 西安近代化学研究所, 陕西 西安 710065; 2. 燃烧与爆炸技术重点实验室, 陕西 西安 710065)
1 引 言
含能材料是武器系统的毁伤威力来源和动力能源,也是制约武器系统应用的关键因素。武器系统的不断发展和进步,对含能材料的性能要求也不断提高,其中高能低感成为近年来含能材料性能所追求的热点[1-4]。理想的含能材料应包括以下特点: 1)高能量; 2)高密度; 3)高爆速、爆压; 4)热稳定性好; 5)低感度。然而,高能量和低感度两方面通常是相互矛盾和对立的。例如, RDX、HMX、CL-20等高能材料使武器装备系统具有较大的毁伤威力,但安全性能下降。而TATB等钝感材料虽然使武器装备系统的安全性能大幅提升,却以损失毁伤威力为代价。因此,如何使高能量和低感度得到更好地融合和兼顾,成为新型含能材料研究领域的热点和挑战[5-7]。
近年来,多硝基唑类化合物在含能材料领域具有良好的应用前景。典型的多硝基唑类化合物主要包括1-甲基-2,4,5-三硝基咪唑(MTNI)、4-氨基-3,5-二硝基吡唑(LLM-116)、3-硝基-1,2,4-三唑-5-酮(NTO)等。此类化合物分子中含有大量键能较高的N—N键、C—N键和致爆基团—NO2,因而具有较高的生成焓和氧平衡; 同时,由于芳杂环的稳定性较好,可以有效增强此类化合物对热、摩擦和撞击的耐受能力[8-11]。三硝甲基唑类化合物具有致爆基团多、能量高、氧平衡高、稳定性高等特点,成为多硝基唑类化合物研究领域的热点之一。例如,2011年,Thottempudi V等[12]报道了两种多硝基三唑类含能化合物5-硝基-3-三硝甲基-1H-1,2,4-三唑(TNNT)和5,5′-双(三硝甲基)-3,3′-偶氮-1H-1,2,4-三唑(BTNAT)的合成及性能,其氧平衡分别为+9.12%和-8.6%,实测密度分别为1.94 g·cm-3和1.83 g·cm-3, 理论爆速(爆压)分别为8983 m·s-1(35.51 GPa)和8964 m·s-1(36.65 GPa),二者的爆轰性能优于TNT,与RDX基本相当,其撞击感度分别为9.0 J和1.5 J,前者较RDX和HMX的撞击感度低,后者较为敏感。此外,由于三硝甲基较强的吸电子作用,二者分子结构中唑环上1位N—H键上的氢原子具有较大的酸性,可以利用其酸性,以其作为阴离子与各种含能阳离子进行配对反应就可以形成具有高能低感特征的离子型含能化合物。此外,还可以通过对1位N—H键上的氢原子进行取代反应和进一步的含能化修饰,从而衍生出能量更高的一系列新型高能化合物。目前,国内尚未开展TNNT和BTNAT的合成研究。因此,本研究根据文献[12]报道的方法合成了TNNT和BTNAT,并对其进行了差示扫描量热(DSC)分析,为其在含能材料中的研究奠定基础。
2 实验部分
2.1 材料及仪器
材料: 氨基胍碳酸氢盐、丙二酸,分析纯,成都市科龙化工试剂厂; 氢氧化钾、高锰酸钾,分析纯,成都市科龙化工试剂厂; 发烟硝酸,自制; 二氯甲烷,分析纯,天津化学试剂三厂。
仪器: NEXUS 870型傅里叶变换红外光谱仪,美国NICOLET公司; AV500型(500 MHz)超导核磁共振波谱仪(TMS内标),德国BRUKER公司; VARIO EL Ⅲ型有机元素分析仪,德国ELEMENTAR公司; 901 s差式扫描量热仪,美国TA公司。
2.2 合成路线
以氨基胍碳酸氢盐与丙二酸为原料,经缩合-环化反应得到中间体5-氨基-1H-1,2,4-三唑-3-乙酸(ATAA); 经重氮化-取代反应得到中间体5-硝基-1H-1,2,4-三唑-3-乙酸(NTAA),进而经硝化反应合成出目标化合物TNNT; 中间体ATAA经氧化偶联反应得到中间体5,5′-双(羧甲基)-3,3′-偶氮-1H-1,2,4-三唑(BCMAT),最后经硝化反应合成出目标化合物BTNAT。合成路线如Scheme 1所示。
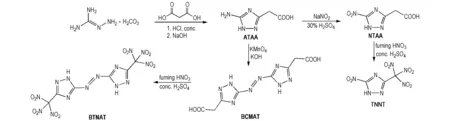
Scheme 1[12]
2.2.1 5-氨基-1H-1,2,4-三唑-3-乙酸(ATAA)
称取30 g(0.2205 mol)氨基胍碳酸氢盐、22.95 g(0.2205 mol)丙二酸加入500 mL三口反应瓶中,搅拌下缓慢滴加25.85 g(0.255 mol)36%的浓盐酸。滴毕,升温至70℃并恒温反应60~70 min。反应完成后冷却至20 ℃,然后向所得粘性反应物中加入95 mL溶有22.58 g(0.5645 mol)氢氧化钠的水溶液,加热至90~95 ℃反应40 min,然后冷却至50~60 ℃。然后用36%的盐酸调节pH为2~3,并冷却至5 ℃; 经过滤、干燥得白色固体; 然后用蒸馏水重结晶,得白色固体粉末14.09 g,产率为45%(文献值[12]: 44%~51%)。
m.p.: 187 ℃; IR(KBr,ν/cm-1) : 3463, 3425, 3338, 3114, 2714, 1686, 1597, 1557, 1516, 1378, 1263, 1194, 1108, 1060, 1005, 919, 826, 759, 682, 645, 587, 536;1H NMR(DMSO-d6,500 MHz)δ: 11.2484 br.s(1H, NH),5.7960 br.s(2H, NH2),3.5915s(2H, CH2);13C NMR(DMSO-d6,125 MHz)δ: 170.953, 157.804, 153.892, 34.469; Anal. Calcd for C4H6N4O2, %: C 33.81,H 4.26,N 39.42; Found, %: C 34.00,H 4.19,N 39.18。
2.2.2 5-硝基-1H-1,2,4-三唑-3-乙酸(NTAA)
称取41.4 g(600 mmol)亚硝酸钠溶于100 mL蒸馏水中并加热至50 ℃,再取8.52 g(60 mmol)5-氨基-1H-1,2,4-三唑-3-乙酸溶于50 mL30%硫酸溶液中,并将所得溶液滴加至亚硝酸钠水溶液中。滴毕,50 ℃恒温反应1 h,然后冷却至25 ℃,用30%硫酸溶液调节pH至1,搅拌至无二氧化氮冒出,然后用乙酸乙酯萃取(100 mL×3),减压蒸除乙酸乙酯得淡黄色固体。将所得固体用蒸馏水重结晶,得4.13 g淡黄色固体粉末,产率为40%(文献值[12]: 42%)。
IR(KBr,ν/cm-1): 3437, 3278, 2979, 2925, 2732, 2482, 1940, 1695, 1562, 1539, 1494, 1438, 1393, 1341, 1314,1268, 1229, 1165, 1051, 1026, 842, 816, 778, 690, 654;1H NMR(DMSO-d6, 500 MHz)δ: 15.1378 br.s(1H, NH),3.9831s(2H, CH2);13C NMR(DMSO-d6,125 MHz)δ: 169.373, 162.856, 153.458, 33.156; Anal. Calcd for C4H4N4O4, %: C 27.92,H 2.34,N 32.55; Found, %: C 27.90, H 2.29, N 31.99。
2.2.3 5-硝基-3-三硝甲基-1,2,4-三唑(TNNT)
量取4 mL发烟硝酸和5 mL 98%浓硫酸加入25 mL反应瓶中,冰盐浴冷却至0 ℃,缓慢、分批加入1 g(5.8 mmol)5-硝基-1H-1,2,4-三唑-3-乙酸。加毕,室温下反应15 h。反应完成后将所得反应混合物倒入15 g冰水中,用30%氢氧化钠溶液调节混合液酸碱性至pH=8,再用浓盐酸调节混合液酸碱性至pH=2~3; 然后用二氯甲烷萃取(10 mL×3),无水硫酸镁干燥萃取液,经减压蒸除二氯甲烷得无色固体0.67 g,产率为42%(文献值[12]: 44%)。
IR(KBr,ν/cm-1): 3444, 2960, 2924, 2853, 1705, 1600, 1570, 1465, 1379, 1283, 1261, 1088, 1020, 844, 801 ;1H NMR(CD3CN,500 MHz)δ: 9.401 s;13C NMR(CD3CN,125 MHz)δ: 156.64, 146.11, 121.01;15N NMR(CD3CN)δ: -37.58, -89.89, -131.51, -143.74, -146.15; Anal. Calcd for C3HN7O8, %: C 13.70, H 0.38, N 37.27; Found, %: C 14.18, H 0.30, N 37.52。
2.2.4 5,5′-双(羧甲基)-3,3′-偶氮-1H-1,2,4-三唑(BXMAT)
称取8.52 g(60 mmol)5-氨基-1H-1,2,4-三唑-3-乙酸、40 mL蒸馏水加入到250 mL三口反应瓶中,搅拌形成悬浮液,然后滴加40 mL溶有12 g(214 mmol)氢氧化钾的水溶液,滴加过程持续30~40 min; 待反应体系变为透明后分批加入8 g(50 mmol)高锰酸钾。加毕,在25~30 ℃下反应2 h; 反应完成后过滤并收集滤液,用36%盐酸调节pH至1~1.5,析出黄色固体,经过滤、真空干燥得黄色固体粉末14.29 g,产率为85%(文献值[12]: 84%)。
IR(KBr,ν/cm-1) : 3467, 2994, 2923, 1623, 1596, 1541, 1446, 1372, 1283, 1190, 1095, 1043, 996, 963, 948, 843, 801 ;1H NMR(DMSO,500 MHz)δ: 14.7364 br.s(2H, NH), 3.9126 s(4H, CH2), 3.3965 br.s(2H, COOH);13C NMR(DMSO,125 MHz)δ: 169.498, 168.543, 152.375, 33.009; Anal. Calcd for C8H8N8O4, %: C 34.29,H 2.88,N 39.99; Found, %: C 34.17, H 2.32, N 39.92。
2.2.5 5,5′-双(三硝甲基)-3,3′-偶氮-1,2,4-三唑(BTNAT)
量取12 mL 98%浓硫酸和10 mL发烟硝酸加入50 mL反应瓶中,冰浴冷却至-5~0 ℃,缓慢、分批加入1 g(3.57 mmol) 5,5′-双(羧甲基)-3,3′-偶氮-1H-1,2,4-三唑。加毕,缓慢升至室温,并继续反应15 h。反应完成后,将所得反应混合物投入30 g冰水中,用30%氢氧化钠溶液调节混合液酸碱性至pH=8,再用浓盐酸调节混合液酸碱性至pH=2~3; 过滤得黄色粉末状固体; 收集滤液,用乙酸乙酯萃取并无水硫酸镁干燥,最后减压蒸除乙酸乙酯得黄色粉末状固体。将两次所得黄色固体合并,共计0.693 g,产率为42%(文献值[12]: 55%)。
IR(KBr,ν/cm-1) : 3443, 3125, 2994, 2923, 1743, 1623, 1596, 1541, 1446, 1372, 1283, 1241, 1190, 1095, 996, 948, 843, 801;1H NMR(DMSO,500 MHz)δ: 5.062 s;13C NMR(DMSO,125 MHz)δ: 165.33, 149.15, 138.216; Anal. Calcd for C6H2N14O12, %: C 15.59, H 0.44, N 42.43; Found, %: C 15.42, H 0.39, N 41.96。
3 结果与讨论
3.1 硝化反应体系的选择
据文献[13]报道,唑环上的乙酸基或乙酸乙酯基在硝酸或硝硫混酸体系中经硝化反应可转化为三硝甲基结构。因此,本研究分别考察了不同硝化反应前体和硝化剂种类对两种硝化反应的影响。本研究选择四种结构相似的三唑类化合物即ATAA、5-氨基-1H-1,2,4-三唑-3-乙酯(ATEE)、5-硝基-1H-1,2,4-三唑-3-乙酸(NTAA)和5-硝基-1H-1,2,4-三唑-3-乙酯(NTEE)作为合成TNNT的前体(见Scheme 2),其中ATEE和NTEE的合成方法可参照文献[14]。同样,选择BCMAT和5,5′-双(甲基乙酯基)-3,3′-偶氮-1H-1,2,4-三唑(BCMET)作为合成BTNAT的前体(见Scheme 3),其中BCMET的合成方法可参照文献[15]。
同时,选择工业硝酸、发烟硝酸和四种不同配比的硝硫混酸作为硝化剂,实验结果见表1。
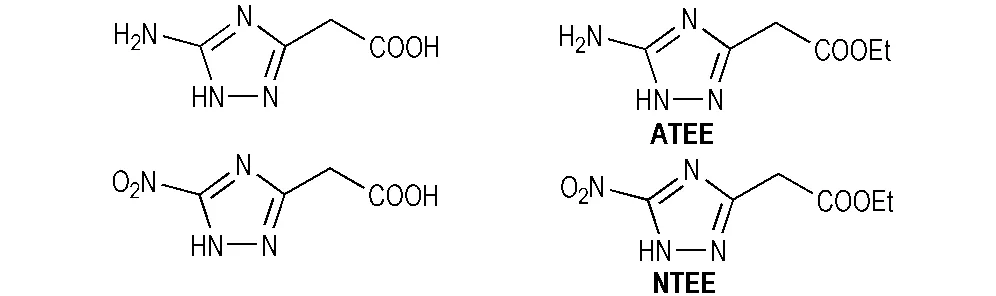
Scheme 2
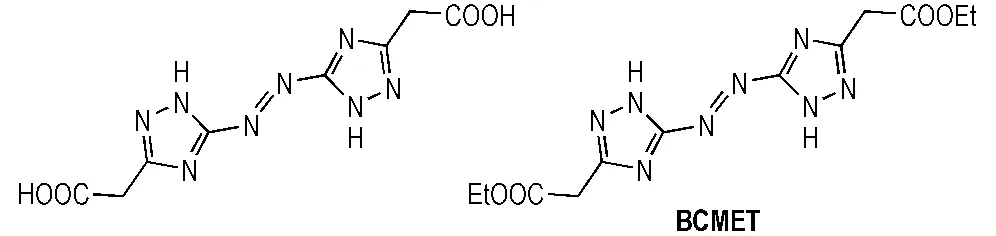
Scheme 3
由表1可见,ATAA和ATEE分别属于氨基取代的三唑类化合物,无论采用何种硝化剂,得到的都是混合物,且难以分离。究其原因,是由于硝化反应不仅可以发生在乙酸基或乙酸乙酯基的仲碳位置上,而且5C位的氨基活性较高,且易硝化生成N-硝化副产物。当ATAA和ATEE分别经重氮化-取代反应转化为硝基取代的三唑类化合物时,由于排除了N-硝化副反应的干扰,可以顺利得到目标化合物; 此外,由表1可以看出,以NTEE作为硝化前体的反应收率明显低于NTAA,这是因为乙酸乙酯基在硝化反应过程中脱羧能力较弱,从而生成大量的偕二硝甲基取代物和少量的三硝甲基取代物; 而以NTAA作为硝化前体时,乙酸基在反应过程中更容易脱去羧基而继续被硝化,从而得到的只有三硝甲基取代物。NTEE和NTAA的硝化反应式如Scheme 4所示。
表1 不同硝化体系对硝化反应的影响
Table 1 Effect of different nitration system on the nitration reaction

nitrationprecursoryield/%98%nitricacid100%nitricacidmixedacid[V(98%H2SO4)/V(100%HNO3)]1∶11.2∶11.5∶12∶1ATAAoilmixturedoesnotseparateNTAA<51242443520ATEEoilmixturedoesnotseparateNTEE<51022282017BCMAT<52233424030BCMET<51825383222
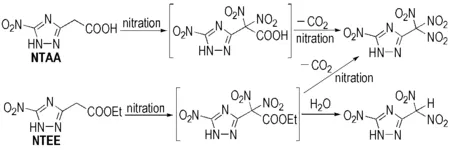
Scheme 4
同理,Scheme 4所示机理同样适用于BCMAT和BCMET的硝化体系,在此不再赘述。因此,以NTAA和BCMAT分别为硝化前体,硝硫混酸[V(98% H2SO4)/V(100% HNO3)=1.2/1]作为硝化剂是最合适的硝化体系。
3.2 后处理方式研究
实验过程中发现,如果完全按照文献[12]报道的实验步骤进行后处理,得到的产物经红外分析可发现明显的成盐特征,原因在于三唑环结构上的1位N—H键在强酸环境中可显弱碱性,能够与硝酸或硫酸发生反应从而形成离子盐结构,导致产物纯度不高。因此,本研究在后处理过程中增加了破坏离子盐结构的操作步骤,即首先用碱对反应混合物进行酸碱性调节至体系pH=8,然后再进行酸化,进而得到较为纯净的目标化合物。此外,这种后处理方式也是造成本研究产率较文献报道值低的原因。
3.3 量子化学计算
3.3.1 几何优化
采用密度泛函理论(DFT)的B3LYP方法[16-17],在6-31G(d,p)基组水平上对TNNT和BTNAT的结构进行了全优化,经振动频率分析发现无虚频,表明优化结构为势能面上的极小点,为稳定构型。优化后的几何构型及原子编号见图1。键长、键角和二面角数据分别见表2和表3。
由表2和表3可见,TNNT和BTNAT三唑环上的C—N键的键长均接近C—N、CN和N—N键的标准键长(1.4720Å、1.2870Å和1.450Å),且各键长呈现出平均化,表明三唑环结构形成了共轭体系; 三硝甲基单元中三个C—N键的键长较C—N键的标准键长要长,表明其键能较低,相对较弱。
3.3.2 自然键轨道分析及键离解能
政府补助。政府补助主要包括财政贴息、研究开发补贴和政策性补贴。该变量的数据来源于企业财务报表附注中损益项目下政府补助项目,是当期企业收到的政府补助合计减去退回政府补助的金额。
对TNNT和BTNAT的优化构型进行自然键轨道(NBO)分析,所得C、H、N和O之间的键级以及热引发键的键离解能分别列于表4和表5中。
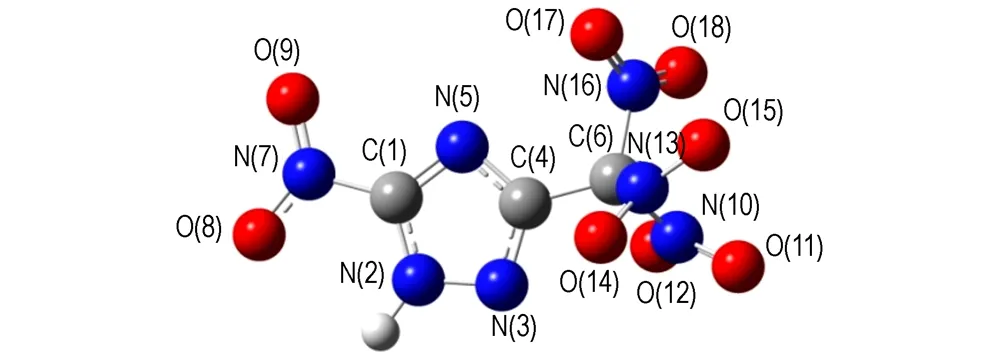
a. TNNT
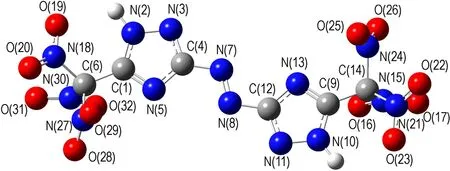
b. BTNAT
图1 优化后TNNT和BTNAT的几何构型
Fig.1 The geometric configurations of TNNT and BTNAT after optimization
表2 TNNT的部分几何参数
Table 2 Selected geometric parameters for TNNT
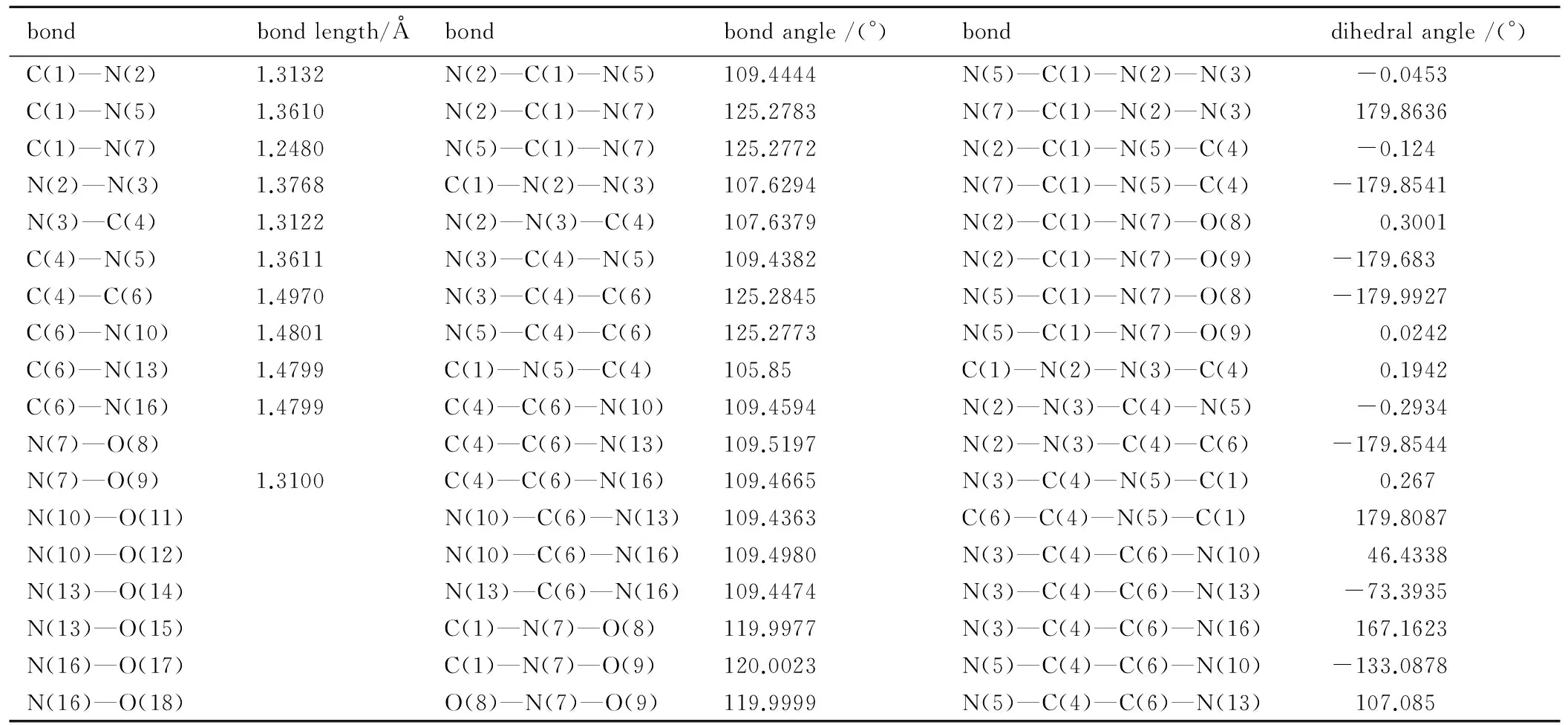
bondbondlength/Åbondbondangle/(°)bonddihedralangle/(°)C(1)—N(2)1.3132N(2)—C(1)—N(5)109.4444N(5)—C(1)—N(2)—N(3) -0.0453C(1)—N(5)1.3610N(2)—C(1)—N(7)125.2783N(7)—C(1)—N(2)—N(3) 179.8636C(1)—N(7)1.2480N(5)—C(1)—N(7)125.2772N(2)—C(1)—N(5)—C(4) -0.124N(2)—N(3)1.3768C(1)—N(2)—N(3)107.6294N(7)—C(1)—N(5)—C(4) -179.8541N(3)—C(4)1.3122N(2)—N(3)—C(4)107.6379N(2)—C(1)—N(7)—O(8) 0.3001C(4)—N(5)1.3611N(3)—C(4)—N(5)109.4382N(2)—C(1)—N(7)—O(9) -179.683C(4)—C(6)1.4970N(3)—C(4)—C(6)125.2845N(5)—C(1)—N(7)—O(8) -179.9927C(6)—N(10)1.4801N(5)—C(4)—C(6)125.2773N(5)—C(1)—N(7)—O(9) 0.0242C(6)—N(13)1.4799C(1)—N(5)—C(4)105.85C(1)—N(2)—N(3)—C(4) 0.1942C(6)—N(16)1.4799C(4)—C(6)—N(10)109.4594N(2)—N(3)—C(4)—N(5) -0.2934N(7)—O(8)C(4)—C(6)—N(13)109.5197N(2)—N(3)—C(4)—C(6) -179.8544N(7)—O(9)1.3100C(4)—C(6)—N(16)109.4665N(3)—C(4)—N(5)—C(1) 0.267N(10)—O(11)N(10)—C(6)—N(13)109.4363C(6)—C(4)—N(5)—C(1) 179.8087N(10)—O(12)N(10)—C(6)—N(16)109.4980N(3)—C(4)—C(6)—N(10) 46.4338N(13)—O(14)N(13)—C(6)—N(16)109.4474N(3)—C(4)—C(6)—N(13) -73.3935N(13)—O(15)C(1)—N(7)—O(8)119.9977N(3)—C(4)—C(6)—N(16) 167.1623N(16)—O(17)C(1)—N(7)—O(9)120.0023N(5)—C(4)—C(6)—N(10) -133.0878N(16)—O(18)O(8)—N(7)—O(9)119.9999N(5)—C(4)—C(6)—N(13) 107.085
表3 BTNAT的部分几何参数
Table 3 Selected geometric parameters for BTNAT
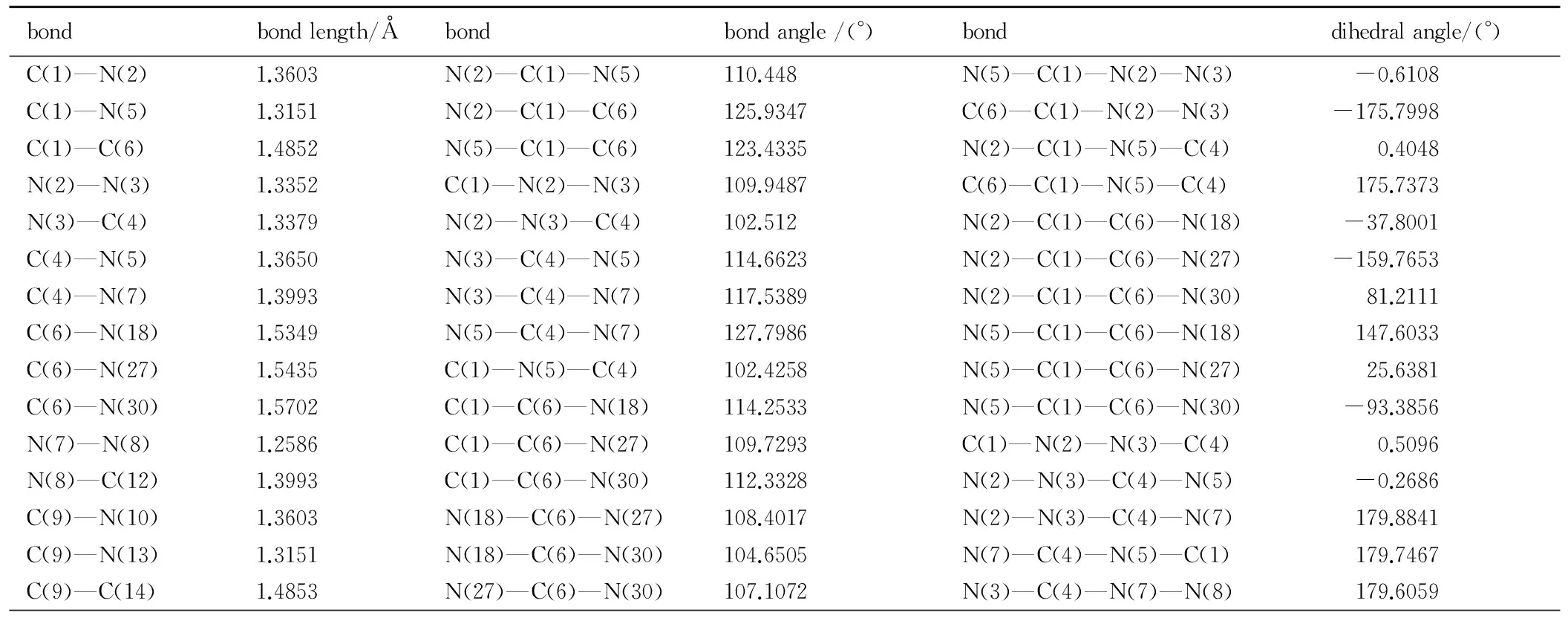
bondbondlength/Åbondbondangle/(°)bonddihedralangle/(°)C(1)—N(2)1.3603N(2)—C(1)—N(5)110.448N(5)—C(1)—N(2)—N(3) -0.6108C(1)—N(5)1.3151N(2)—C(1)—C(6)125.9347C(6)—C(1)—N(2)—N(3) -175.7998C(1)—C(6)1.4852N(5)—C(1)—C(6)123.4335N(2)—C(1)—N(5)—C(4) 0.4048N(2)—N(3)1.3352C(1)—N(2)—N(3)109.9487C(6)—C(1)—N(5)—C(4) 175.7373N(3)—C(4)1.3379N(2)—N(3)—C(4)102.512N(2)—C(1)—C(6)—N(18) -37.8001C(4)—N(5)1.3650N(3)—C(4)—N(5)114.6623N(2)—C(1)—C(6)—N(27) -159.7653C(4)—N(7)1.3993N(3)—C(4)—N(7)117.5389N(2)—C(1)—C(6)—N(30) 81.2111C(6)—N(18)1.5349N(5)—C(4)—N(7)127.7986N(5)—C(1)—C(6)—N(18) 147.6033C(6)—N(27)1.5435C(1)—N(5)—C(4)102.4258N(5)—C(1)—C(6)—N(27) 25.6381C(6)—N(30)1.5702C(1)—C(6)—N(18)114.2533N(5)—C(1)—C(6)—N(30) -93.3856N(7)—N(8)1.2586C(1)—C(6)—N(27)109.7293C(1)—N(2)—N(3)—C(4) 0.5096N(8)—C(12)1.3993C(1)—C(6)—N(30)112.3328N(2)—N(3)—C(4)—N(5) -0.2686C(9)—N(10)1.3603N(18)—C(6)—N(27)108.4017N(2)—N(3)—C(4)—N(7) 179.8841C(9)—N(13)1.3151N(18)—C(6)—N(30)104.6505N(7)—C(4)—N(5)—C(1) 179.7467C(9)—C(14)1.4853N(27)—C(6)—N(30)107.1072N(3)—C(4)—N(7)—N(8) 179.6059
续表1
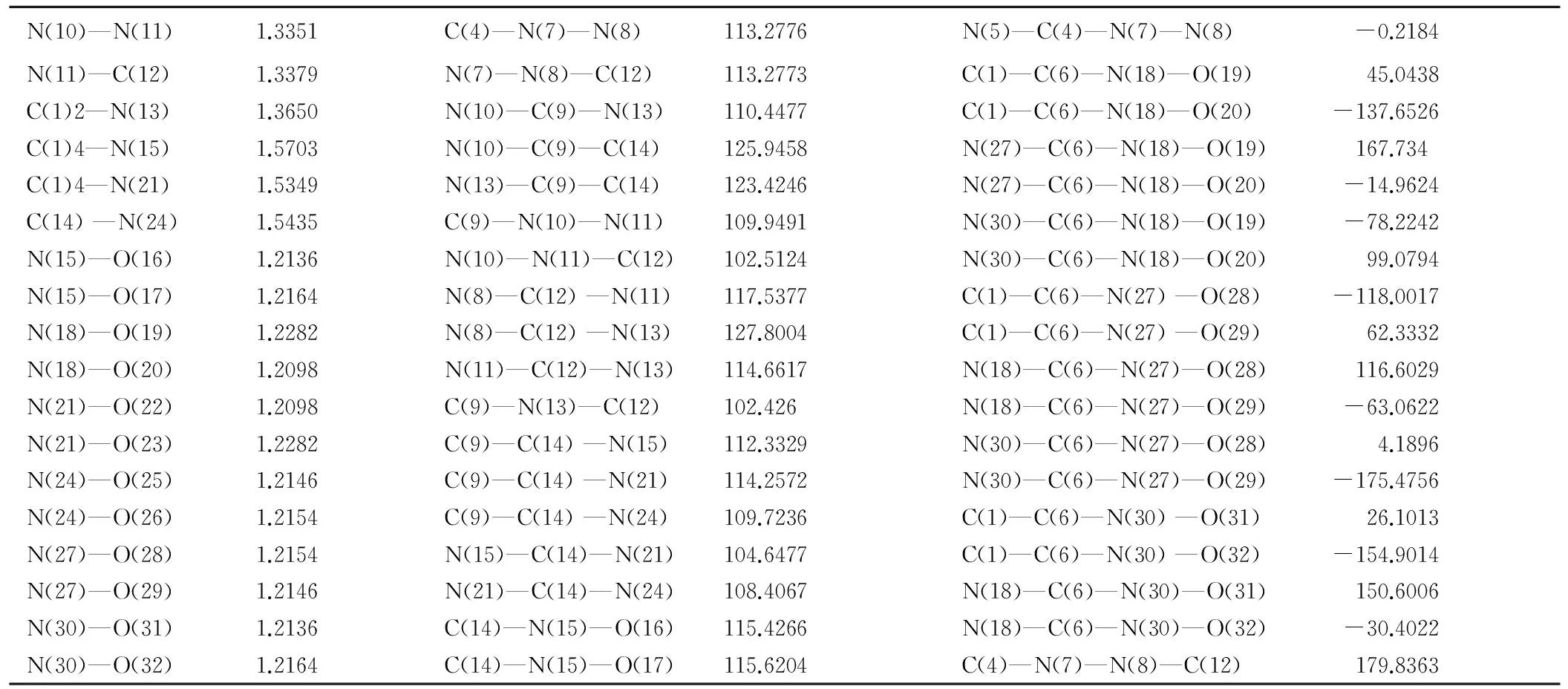
N(10)—N(11)1.3351C(4)—N(7)—N(8)113.2776N(5)—C(4)—N(7)—N(8) -0.2184N(11)—C(12)1.3379N(7)—N(8)—C(12)113.2773C(1)—C(6)—N(18)—O(19) 45.0438C(1)2—N(13)1.3650N(10)—C(9)—N(13)110.4477C(1)—C(6)—N(18)—O(20) -137.6526C(1)4—N(15)1.5703N(10)—C(9)—C(14)125.9458N(27)—C(6)—N(18)—O(19) 167.734C(1)4—N(21)1.5349N(13)—C(9)—C(14)123.4246N(27)—C(6)—N(18)—O(20) -14.9624C(14)—N(24)1.5435C(9)—N(10)—N(11)109.9491N(30)—C(6)—N(18)—O(19) -78.2242N(15)—O(16)1.2136N(10)—N(11)—C(12)102.5124N(30)—C(6)—N(18)—O(20) 99.0794N(15)—O(17)1.2164N(8)—C(12)—N(11)117.5377C(1)—C(6)—N(27)—O(28) -118.0017N(18)—O(19)1.2282N(8)—C(12)—N(13)127.8004C(1)—C(6)—N(27)—O(29) 62.3332N(18)—O(20)1.2098N(11)—C(12)—N(13)114.6617N(18)—C(6)—N(27)—O(28) 116.6029N(21)—O(22)1.2098C(9)—N(13)—C(12)102.426N(18)—C(6)—N(27)—O(29) -63.0622N(21)—O(23)1.2282C(9)—C(14)—N(15)112.3329N(30)—C(6)—N(27)—O(28) 4.1896N(24)—O(25)1.2146C(9)—C(14)—N(21)114.2572N(30)—C(6)—N(27)—O(29) -175.4756N(24)—O(26)1.2154C(9)—C(14)—N(24)109.7236C(1)—C(6)—N(30)—O(31) 26.1013N(27)—O(28)1.2154N(15)—C(14)—N(21)104.6477C(1)—C(6)—N(30)—O(32) -154.9014N(27)—O(29)1.2146N(21)—C(14)—N(24)108.4067N(18)—C(6)—N(30)—O(31)150.6006N(30)—O(31)1.2136C(14)—N(15)—O(16)115.4266N(18)—C(6)—N(30)—O(32) -30.4022N(30)—O(32)1.2164C(14)—N(15)—O(17)115.6204C(4)—N(7)—N(8)—C(12) 179.8363
表4 TNNT的Wiberg键级及键离解能
Table 4 Wiberg bond orders and BDE of TNNT
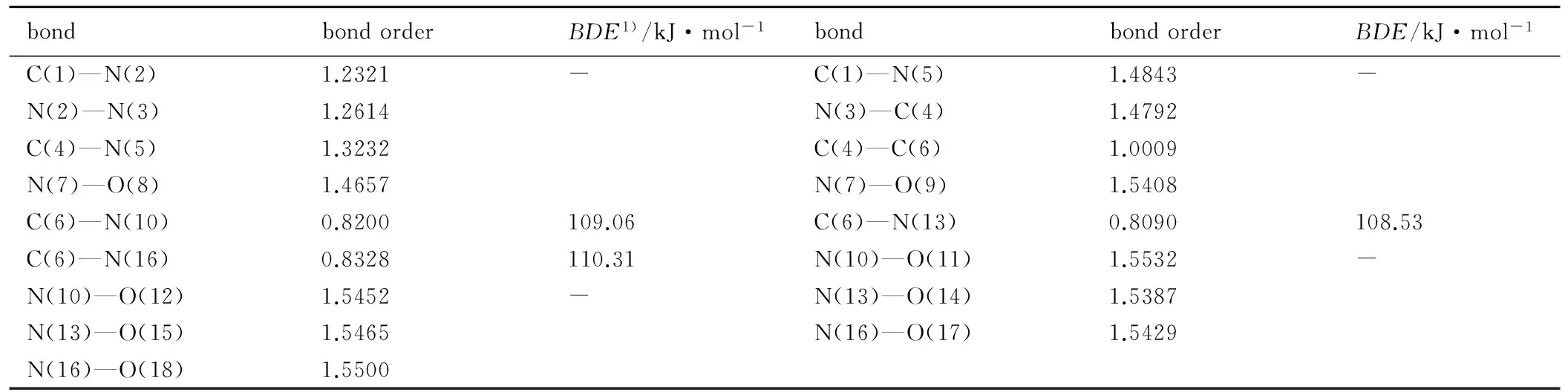
bondbondorderBDE1)/kJ·mol-1bondbondorderBDE/kJ·mol-1C(1)—N(2)1.2321-C(1)—N(5)1.4843-N(2)—N(3)1.2614N(3)—C(4)1.4792C(4)—N(5)1.3232C(4)—C(6)1.0009N(7)—O(8)1.4657N(7)—O(9)1.5408C(6)—N(10)0.8200109.06C(6)—N(13)0.8090108.53C(6)—N(16)0.8328110.31N(10)—O(11)1.5532-N(10)—O(12)1.5452-N(13)—O(14)1.5387N(13)—O(15)1.5465N(16)—O(17)1.5429N(16)—O(18)1.5500
Note: 1)BDEis bond dissociation energy.
表5 BTNAT的Wiberg键级
Table 5 Wiberg bond orders of BTNAT
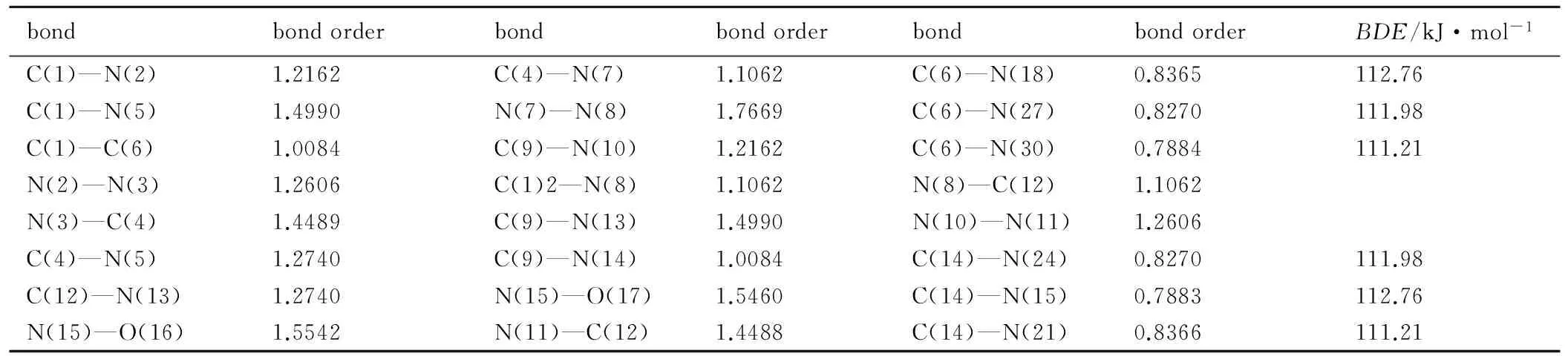
bondbondorderbondbondorderbondbondorderBDE/kJ·mol-1C(1)—N(2)1.2162C(4)—N(7)1.1062C(6)—N(18)0.8365112.76C(1)—N(5)1.4990N(7)—N(8)1.7669C(6)—N(27)0.8270111.98C(1)—C(6)1.0084C(9)—N(10)1.2162C(6)—N(30)0.7884111.21N(2)—N(3)1.2606C(1)2—N(8)1.1062N(8)—C(12)1.1062N(3)—C(4)1.4489C(9)—N(13)1.4990N(10)—N(11)1.2606C(4)—N(5)1.2740C(9)—N(14)1.0084C(14)—N(24)0.8270111.98C(12)—N(13)1.2740N(15)—O(17)1.5460C(14)—N(15)0.7883112.76N(15)—O(16)1.5542N(11)—C(12)1.4488C(14)—N(21)0.8366111.21
由表3可见,TNNT结构中C(6)—N(10)、C(6)—N(13)和C(6)—N(16)的和BTNAT结构中C(6)—N(18)、C(6)—N(27)、C(6)—N(30)、C(14)—N(15)、C(14)—N(21)、C(14)—N(24)键级最小,属于热解引发键,均为三硝甲基中的C—N键,表明三硝甲基唑类化合物不稳定的关键因素为三硝甲基结构中C—NO2的热裂解。随着温度的升高,该键会首先发生断裂,进而引发含能化合物的热分解。此外,还可通过热裂解键的键离解能的大小来判断含能化合物的热稳定性。肖鹤鸣等[18]认为计算所得热解引发键的键离解能(BDE)可作为稳定性的定量标准。若BDE>80 kJ·mol-1,认为达到基本要求; 若BDE>120 kJ·mol-1,认为满足品优高能量密度材料的稳定性要求。由表3可以看出,TNNT和BTNAT结构中三硝甲基上的三个热解引发键的BDE均大于80 kJ·mol-1,基本接近120 kJ·mol-1。故可判断TNNT和BTNAT基本满足高能量密度材料的稳定性要求。
3.3.3 静电势分析
在B3LYP理论水平下,画出了TNNT和BTNAT的三维静电势分布示意图(图2),蓝色部分表示正静电势,红色部分表示负静电势。从图2中可以看出,化合物的正静电势主要分布在三唑环周围,而TNNT中C(1)位所连NO2以及C(4)位所连的三硝甲基和BTNAT中两个三唑之间以及C(1)位、C3位所连的三硝甲基周围属于明显的负电荷区域。通常正电势越强,说明该区域电荷密度较低,故可以看出三唑环上的N原子易受亲核试剂进攻。Anton H等[19]认为在含能体系中,正的静电势强度要大于负的静电势强度。由计算结果可知,TNNT和BTNAT的正静电势区域的静电势强度分别为82.634 kJ·mol-1和67.864 kJ·mol-1,负静电势区域的静电势强度为-34.518 kJ·mol-1和-34.602 kJ·mol-1,符合Anton H等对含能体系的定性判断。
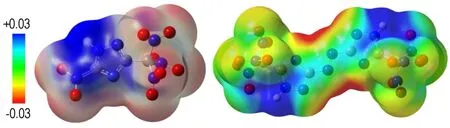
a. TNNT b. BTNAT
图2 TNNT和BTNAT的三维静电势分布示意图
Fig.2 Schematic diagrams of three-dimensional electrostatic potential distribution for TNNT and BTNAT
3.4 热稳定性
升温速率为10 ℃·min-1,氮气气氛,TNNT和BTNAT的DSC曲线如图3所示。
由图3可见,TNNT和BTNAT的DSC曲线没有明显的吸热峰,表明二者在整个温度范围内没有熔化过程; TNNT和BTNAT的分解峰温分别为135 ℃和146 ℃(文献[12]报道值150 ℃)。此外,二者的分解温度均低于TNT(295 ℃)、RDX(230 ℃)和HMX(287 ℃)[12],说明TNNT和BTNAT的热稳定性较差,尚需进行相关改性才能满足含能材料对热稳定性的要求。
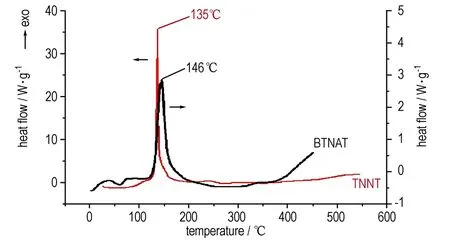
图3 TNNT和BTNAT的DSC曲线
Fig.3 DSC curves of TNNT and BTNAT
4 结 论
(1) 以氨基胍碳酸氢盐和丙二酸为原料,经缩合-环化反应、重氮化-取代反应、氧化偶联反应和硝化反应分别合成出5-硝基-3-三硝甲基-1H-1,2,4-三唑和5,5′-双(三硝甲基)-3,3′-偶氮-1,2,4-三唑,并用红外光谱、核磁共振和元素分析对其进行了结构表征。
(2) 采用密度泛函理论,在B3LYP/6-31G(d, p)基组水平下对5-硝基-3-三硝甲基-1H-1,2,4-三唑和5,5′-双(三硝甲基)-3,3′-偶氮-1,2,4-三唑进行了全构型优化,并在优化构型基础上进行了自然键轨道(NBO)分析。
(3) DSC分析结果表明: 在升温速率为10 ℃·min-1,氮气气氛条件下5-硝基-3-三硝甲基-1H-1,2,4-三唑和5,5′-双(三硝甲基)-3,3′-偶氮-1,2,4-三唑的分解温度分别为135 ℃和146 ℃。
参考文献:
[1] Joo Y H, Shreeve J M. High-density energetic mono- or bis(oxy)-5-nitroiminotetrazoles[J].AngewChemIntEd, 2010, 49: 7320-7323.
[2] Joo Y H, Shreeve J M. Nitroimno-tetrazolates and Oxy-nitroimio-tetrazolates[J].JAmChemSoc, 2010, 132: 15081-15090.
[3] Wang R, Xu H, Guo Y, et al. Bis[3-(5-nitroimino-1,2,4-triazolate)]-based energetic salts: synthesis and promising properties of a new family of high-density insensitive materials[J].JAmChemSoc, 2010, 132: 11904-11905.
[4] Fischer N, Karaghiosoff K, Klapötke T M, et al. New energetic materials featuring tetrazoles and nitramines-synthesis, characterization and properties[J].ZAnorgAllgChem, 2010, 636: 735-749.
[5] Holl G, Klapötke T M, Polborn K, et al. Structure and bonding in 2-diazo-4,6-dinitrophenol(DNNP)[J].PropellantsExpolsPyrotech, 2003, 28: 153-156.
[6] Katrizky A R, Sommen G L, Gromova A V, et al. Synthetic routs towards tetrazolium and triazoium dinitromethylides[J].ChemHeterocyclCompd, 2005, 41: 111-118.
[7] Göbel M, Klapötke T M. Development and testing of Energetic materials: the concept of high densities based on the trinitroethyl functionality[J].AdvFunctMatar, 2009, 19: 347-365.
[8] Puchala A, Belaj F, Bergman J, et al. On the reaction of 3,4-dihydropyrimidones with nitric acid. Preparation and X-ray structure analysis of a stable nitrolic acid[J].JHeterocyclChem, 2001, 38: 1345-1352.
[9] Zeng Z, Gao H, Twamley B, et al. Energetic mono and dibasic 5-dinitromethyltetrazolates: synthesis, properties and particle processing[J].JMaterChem, 2007, 17: 3819-3826.
[10] Thottempudi V, Gao H, Shreeve J M. Trinitromethyl-substituted 5-nitro- or 3-azo-1,2,4-triazoles: synthesis, characterization, and energetic properties[J].JAmChemSoc, 2011, 133: 6464-6471.
[11] Thottempudi V, Kim T K, Chung K H, et al. Synthesis and characterization of some polynitro imidazoles[J].BullKoreanChemSoc, 2009, 30: 2152-2154.
[12] Abdel-Megeed A M, Abdul-Rahaman H M, Alkaramany G H, et al. Design, synthesis and molecular modeling study of acylated 1,2,4-triazole-3-acetates with potential anti-inflammatory activity[J].EurJMedChem, 2009, 44: 117-123.
[13] Alexander A P, Klapötke T M. Synthesis and characterization of 3,3′-bis(dinitromethyl)-5,5′-azo-1H-1,2,4-triazole[J].ZAnorgAllgChem, 2011, 637:1453-1457.
[14] Becke A D. Density-functional thermochemistry. Ⅲ. The role of exact exchange[J].JournalofChemistryPhysics, 1993, 98(7): 5648-5652.
[15] Lee C, Yang W, Parr R G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].PhysicalReviewB:CondensedMatter, 1988, 37: 785-789.
[16] 肖鹤鸣,许晓娟,邱玲. 高能量密度材料的理论设计[M]. 北京:科学出版社,2008: 38.
XIAO He-ming, XU Xiao-juan, QIU Ling. Theoretical design for high energetic materials[M]. Beijing: Science Press, 2008:38.
[17] Anton H, Klapötke T M, Noth H, et al. Synthesis, structure, molecular orbital and valence bond calculations for tetrazole azide, CHN(7)[J].Propellants,Expolsives,Pyrotechnics, 2003, 28(4): 165-173.
猜你喜欢
化工管理(2021年7期)2021-05-13
化工管理(2020年1期)2020-01-16
中国农资(2016年1期)2016-12-01
中国塑料(2016年11期)2016-04-16
中国果菜(2016年9期)2016-03-01
中国粮油学报(2016年1期)2016-02-06
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
