
牛奶中头孢类药物残留的HPLC多联检测方法
2016-04-27 12:01
中国乳品工业 2016年2期
(山东世通检测评价技术服务有限公司,山东青岛266000;青岛世通检测技术研究院,山东青岛266000)
牛奶中头孢类药物残留的HPLC多联检测方法
许娜,孙海新,张慧,黄金发,孙丕春
(山东世通检测评价技术服务有限公司,山东青岛266000;青岛世通检测技术研究院,山东青岛266000)
采用高效液相色谱法(HPLC),建立了可同时检测牛奶中头孢哌酮、头孢噻肟、头孢曲松、头孢噻吩药物残留量的多联检测方法。样品经超声波振荡,先后加入乙腈除蛋白,正己烷去除脂肪,经0.45μm和0.22μm有机超滤膜过滤,使用Agilent-C18反相色谱柱,以甲醇和0.1%甲酸溶液为流动相(V∶V=4∶6),流速为1m L/min,柱温35℃,检测波长为254 nm。该方法对头孢哌酮、头孢噻肟、头孢曲松、头孢噻吩4种头孢类药物的检出限均为1.0μg/kg,在质量浓度为1.0~50.0μg/L范围内具有良好线性关系,相关系数达到0.999,回收率为90%~105%,批内相对标准偏差≤15%,批间相对标准偏差≤20%。本方法具有分析时间短、检测限低、精密度高、多联检测的优点,对于准确监控牛奶中头孢类药物残留量具有重要意义。
高效液相色谱;头孢;牛奶;药物残留;多联检测
0 引 言
头孢类抗生素(Cephalosporins)药物是临床上用于治疗细菌感染所导致疾病的常用药物,属于β-内酰胺类抗生素[1],具有耐青霉素酶、疗效高、毒性低、过敏反应少、抗菌谱广等特点[2-4],可有效抑制细菌的生长和繁殖。头孢类抗生素在畜牧动物养殖中广泛用于奶牛乳房炎的预防与治疗[5],动物尿道、胃肠道及呼吸道等的细菌病的治疗和防治[6,7]。但由于其使用方法不当或不遵守休药期规定等多方面原因[8,9],引发超剂量用药的恶性循环,给人类带来过敏、肠道菌群失调、耐药性上升等严重危害[10]。因此,建立一种准确、快速的定量检测方法对于实现该类药物的有效监控具有重要意义。
1 实 验
1.1 仪器
Agilent1100高效液相色谱仪,Agilent-C 18(4.6 mm×250mm,5μm)ZORBAX Eclipse Plus反相色谱柱,Cence/湘仪L500台式自动平衡离心机,XW-80A漩涡混合器,CP225D电子天平,UGC-12C旋转式水浴氮吹仪。
1.2 试剂
头孢哌酮(编号130420,纯度>94.7%)、头孢噻肟(编号130483,纯度>93.7%)、头孢曲松(编号130480,纯度>91.8%)、头孢噻吩(编号130407,纯度>96.3%),以上标准品;甲醇、甲酸、乙腈、正己烷(均为色谱纯);水为超纯水;牛奶(全脂灭菌纯牛乳)。
2 方 法
2.1 标准溶液的配置
混合标准品储备溶液:分别准确称取头孢哌酮、头孢噻肟、头孢曲松、头孢噻吩标准品各10 m g,用甲醇-质量分数0.1%甲酸溶解并分别定容到4个100 m L棕色容量瓶中,配制成制成质量浓度为100 m g/L单标准品储备溶液。再分别吸取适量单标准品储备溶液转移至1个100 m L棕色容量瓶中精确定容,配制成质量浓度为1 m g/L的混合标准品储备溶液,备用。
混合标准工作液制备:用甲醇-质量分数0.1%甲酸溶液将混合标准品溶液经适当稀释,配制成质量浓度分别为1.0,5.0,10.0,20.0,50.0μg/L的混合标准品工作液,备用。
2.2 样品前处理
称取5 g液态牛奶样品置于离心管中(精确至0.01 g),加入15~20 m L乙腈,超声波振荡(超声5 s,间隔5 s,60个循环),5 000 r/m in离心10 m in,取上清液,吸取上清液至试管中,氮气吹干,加入等体积甲醇-质量分数0.1%甲酸溶液复溶,加入2~2.5 m L正己烷除脂,振荡,5 000 r/m in离心10 m in,弃去正己烷,先后经0.45μm和0.22μm滤膜过滤后转移至棕色瓶中,备用。
2.3 色谱条件
色谱柱:Agilent(C 18,4.6 mm×250 mm,5μm) ZORBAX Eclipse Plus反相色谱柱。
流动相:A-甲醇,B-0.1%甲酸溶液,A∶B(体积比)=4∶6。
流速为1 m L/m in;检测波长为254 nm;柱温为35℃;进样量为20μL。
2.4 标准曲线绘制
将质量浓度分别为1.0,5.0,10.0,20.0,50.0μg/L的混合标准品工作液,采用高效液相色谱仪进行分析,每一水平设5个重复。以4种头孢类药物的峰面积平均值为纵坐标,质量浓度为横坐标绘制标准曲线。
2.5 结果计算
头孢类药物残留量:

式中:X为试样中头孢类抗生素的残留量,m g/ kg;C为试样溶液中头孢类抗生素的质量浓度,μg/ m L;V为最终样液定容体积,m L;f为稀释倍数;m为试样量,g。(计算结果需扣除空白值。测定结果用两次平行测定的算术平均值表示,保留3位有效数字。)
3 结果与分析
3.1 方法线性方程和检出限
采用HPLC测定牛奶中头孢类抗生素的残留量,流动相使用甲醇和0.1%甲酸溶液,配比为4∶6。结果显示,样品全部分析时间为12 m in,其中头孢哌酮、头孢噻肟、头孢曲松在5 m in之内全部出峰,并且4种药物可以完全分开(4种药物标准溶液色谱图详见图1)该方法在质量浓度为1.0~50.0μg/L范围内具有良好线性关系(如图2~图5所示),相关系数达到0.999。其头孢哌酮、头孢噻肟、头孢曲松和头孢噻吩线性方程、相关系数如表1所示。
采用空白样品加目标化合物的测定方式,以信噪比S/N≥3所对应的浓度确定检测限,以S/N≥10所对应的浓度确定定量限[11],按照方法2.1进行处理,头孢哌酮、头孢噻肟、头孢曲松、头孢噻吩4种头孢类药物的最低检出限(S/N=3)均为1.0μg/kg,定限量(S/ N=10)均为2.0μg/kg。

表1 各化合物检测结果
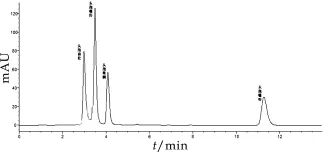
图1 头孢哌酮、头孢噻肟、头孢曲松和头孢噻吩标准溶液色谱
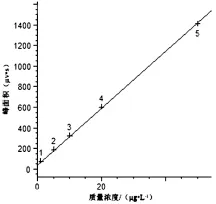
图2 头孢哌酮的标准曲线
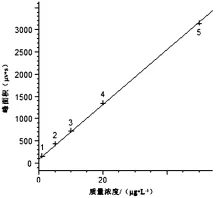
图3 头孢噻肟的标准曲线
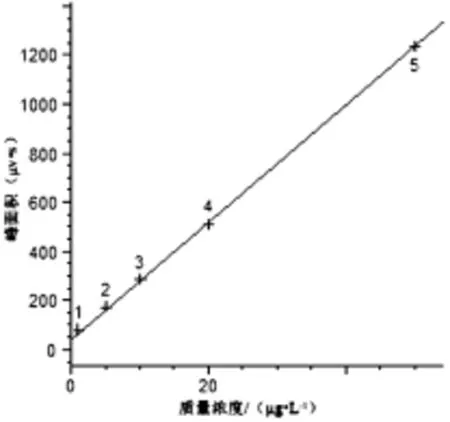
图4 头孢曲松的标准曲线
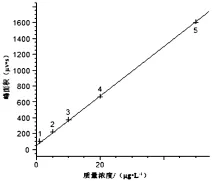
图5 头孢噻吩的标准曲线
3.2 准确度的测定
准确度是指测定值与真实值间的符合程度[12],本研究采用样品添加回收实验,以及高效液相色谱仪分析的结果来反映方法的准确度。本方法的回收率的测定是运用在空白的牛奶样品中添加不同质量分数水平的标准品,进行样品的预处理,采用HPLC方法进行检测,计算回收率。在质量分数为20.0μg/kg水平上添加4种头孢类药物,按本方法测定并计算回收率,每个添加水平重复测定3次,计算批内和批间变异情况,结果如表2所示。由表2可以看出,4种头孢类药物的回收率均在90%~105%之间,批内和批间RSD均小于15%。

表2 牛奶中头孢类药物加标回收率和精密度的测定结果(n=3)
4 讨 论
在整个检测体系的仪器选择方面,本研究采用了高效液相色谱的方式,这是充分考虑到了高效液相色谱方法的稳定性、经济性和高效性[13],相对于更加昂贵和专业的液质联用方法,更易于为广大中小型乳制品生产企业的管理者和技术人员所接收。
样品前处理是各类药物残留检测都无法回避的环节,对于检测体系的稳定性、准确性、灵敏度以及基质效应消除等方面都具有重要的影响[14]。在如乳制品的样品前处理上,Tahira等[12]采用甲醇-质量分数为0.05%甲酸提取和SPE萃取净化,采用2次高速离心使蛋白沉淀等杂质沉淀,但结果表明样品测定结果仍不能消除基质效应影响。Abdalla等[4]用乙酸乙酯提取,甲醇萃取及梯度洗脱,结果仍有几个很强的基质干扰峰。本研究中为了解决基质干扰的问题,综合考虑样品基质里含有的丰富蛋白质和脂肪等物质的影响,以及药物残留在其中的生物结合特点[15]。在解决药物残留与生物基质的结合性方面,采用了间歇性超声波振荡(超声5 s,间隔5 s,60个循环)的方式,通过超声波破坏药物残留与基质的结合,这对于提高准确度具有重要作用[16];在解决蛋白质和脂肪的基质干扰作用方面,本研究通过加入乙腈去除蛋白,加入正己烷去除脂肪,并先后经0.45μm和0.22μm有机超滤膜过滤的方式进行解决,结果表明有效地避免了基质效应的干扰,有效分开个目标待测物。相比于一般样品前处理中采用的SPE柱萃取的方式,本研究所采用的连续过超滤膜的方式更易于操作,节省物力和时间,对于提高检测效率具有积极意义。
5 展 望
本研究已建立相关的样品前处理程序和检测参数优化,更需要进一步形成标准化操作规程,以适应乳制品的质量控制需要,另一方面,考虑到乳制品产品类型的日益多样化,本研究所建立检测技术体系的产业化推广还需要在奶粉类产品、酸奶制品等常见产品中进行进一步的熟化,这需要在今后的工作中作进一步的完善。
[1]PARTA M.Impact of adopting minimum inhibitory concentration as the determinant of susceptibility to cephalosporins and carbapenems in multi-drug resistant Enterobacteriaceae[J].European Journal of Clinical Microbiology&Infectious Diseases,2012,31(6):975-80.
[2]SERGE K.Investigation of the pharmacokinetics and determination of certain cephalosporins in rabbit plasma by a kinetic spectrophotometric method with the aid of chemometrics[J].2011,
[3]CHO K J,KIM JK,LEE J-H,etal.Structural features of cephalosporin acylase reveal the basis of autocatalytic activation[J].Biochemical and Biophysical Research Communications,2009,390(2):342-8.
[4]ELBASHIR A A,AHMED S M A,ABOUL-ENEIN H Y.New spectrofluorimetric method for determination of cephalosporins in pharmaceutical formulations[J].Journal of Fluorescence,2012,22(3): 857-64.
[5]SAMANIDOU V F,TSOCHATZIS E D,PAPADOYANNIS IN. HPLC determination of cefotaxime andcephalexine residues in milk and cephalexine in veterinary formulatio [J].Microchimica Acta,2008,160(4):471-5.
[6]NEUNER E A,R ITCH IE D J,MICEK S T.New antibiotics for healthcare-associated pneumonia;proceedings of the Seminars in respiratory and critical caremedicine,F,2009[C].
[7]NEMUTLU E,KIR S,KATLAN D,et al.Simultaneous multiresponse optimization of an HPLC method to separate seven cephalosporins in plasma and amniotic fluid:Application to validation and quantification of cefepime,cefixim e and cefoperazone[J].Talanta, 2009,80(1):117-26.
[8]GLINKA T,HUIE K,CHO A,et al.Relationships between structure,antibacterial activity,serum stability,pharmacokineticsand efficacy in 3-(heteroarylthio)cephem s.D iscovery of RW J-333441 (MC-04,546)[J].Bioorganic&M edicinal Chem istry,2003,11(4): 591-600.
[9]HECKER SJ,CALKINST,PRICEM E,etal.Prodrugsof cephalosporin RWJ-333441(MC-04,546)with improved aqueous solubility [J].Antim icrobialAgentsand Chemotherapy,2003,47(6):2043-6.
[10]TODA A,OHKIH,YAMANAKA T,et al.Synthesis and SAR of
novel parenteral anti-pseudomonal cephalosporins:discovery of FR 264205[J].Bioorganic&Medicinal Chemistry Letters,2008,18 (17):4849-52.
[11]KAC M,MARSEL J,POKORNY M.On the Analysisof Fermentation Broth during Biosynthesis of Cephalosporin Antibiotics[M]. Springer,1979.
[12]QURESH I T,MEMON N,MEMON SQ,et al.LC/UV determination of cefradine,cefuroxime,and cefotaxime in dairymilk,human serum and wastewater samples[J].SpringerPlus,2013,2(1):575.
[13]MILLER R,AFFOLDER C,NEUSSN.HPLC of cephalosporins and their oxa-derivatives[J].Experientia,1981,37(9):928-30.
[14]王玉堂,吕永辉.目前允许使用的渔药构成与释析[J].中国水产, 2008,1):61-7.
[15]ENGALYTCHEFF A,VANHELLEPUTTE J-P,TILQU IN B.HPLC detection and quantification of radiolytic products of eightβ-blockers irradiated in the solid state and hypotheses on their origins[J]. Pharmaceutical Research,2004,21(7):1103-9.
[16]SANLI S,SAN LI N,GUMUSTAS M,et al.Simultaneous estimation of ceftazidime and ceftizoxime in pharmaceutical formulations by HPLC method[J].Chromatographia,2011,74(7-8):549-58.
High performance liquid chromatography(HPLC)multigang determination method of cephalosporins in milk
XU Na,SUN Hai-xin,ZHANG Hui,HUANG Jin-fa,SUN Pi-chun
(Shandong Seatone Detection Evaluation Co.Ltd.,Qingdao 266000,China;Qingdao Seatone Detection Technology Institute,Qingdao 266000,China)
A HPLC method was established for the multi project joint determination of four cephalosporins(cefoperazone,cefotaxime,ceftriaxone,cefalotin)in milk.The milk sample was deproteinized by acetonitrile and degrease by hexane with ultrasonic technique,then filtered by organic-inorganic hybrid ultrafiltration membranes and detectd under the condition of Agilent-C18 reversed phase chromatographic column,with methanol solution and 0.1%phthalatesle solution(V∶V=4∶6)as mobile phase,flow rates1m L/m in,column temperature as 35℃, test wavelength as254 nm.The results showed that the four cephalosporins were good linear from 1~50μg/L,with minimum limits of detection were 1.0μg/kg respectively,and the correlation coefficients r2were greater than 0.999,the recoveries were at the levels90%~105%, intra-batch and inter-batch RSD were less than 15%and 20%respectively.The established multi project joint method can be applied for the determination of cephalosporins residue in milk within a short span of time,high minimum limits of detection,and high precision.
high performance liquid chromatography(HPLC);cephalosporin;milk;drug residue;multy determination
TS252.7
:A
:1001-2230(2016)02-0050-03
2015-09-07
国家火炬计划项目(2015GH581533);青岛市技术创新平台建设计划项目(14-7-2-36-gx)。
许娜(1986-),女,工程师,研究方向为食品安全检测与质量控制研究。
孙海新
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10
中国药学药品知识仓库(2022年7期)2022-05-10
健康体检与管理(2022年2期)2022-04-15
保健与生活(2021年16期)2021-08-16
昆明医科大学学报(2021年6期)2021-07-31
医学食疗与健康(2021年27期)2021-05-13
中华养生保健(2020年2期)2020-11-16
中国抗生素杂志(2019年12期)2019-12-30
中国现代药物应用(2015年5期)2015-03-07
中国当代医药(2015年7期)2015-03-01
