
人类蛋白组学草图的肺癌分子标记物初探
2016-04-26 09:29朱琰琰罗执芬崔永霞卢创新
生物信息学 2016年1期
朱琰琰,罗执芬,崔永霞,卢创新,周 云*
(1.河南省人民医院肿瘤科, 郑州 450003;
2.郑州大学人民医院肿瘤科, 郑州 450003)
人类蛋白组学草图的肺癌分子标记物初探
朱琰琰1,2,罗执芬1,2,崔永霞1,2,卢创新1,2,周云1,2*
(1.河南省人民医院肿瘤科, 郑州 450003;
2.郑州大学人民医院肿瘤科, 郑州 450003)
摘要:传统的肺癌分子标记物探索通常基于基因组或者转录组研究,而基于蛋白质水平的肺癌分子标记物探索通常局限在低通量水平。 质谱技术已经开始产生高通量的全局正常及癌症蛋白组。我们采用开源统计软件R对人类蛋白组学草图数据及已发表的肺癌蛋白质组学数据进行二次分析,筛选出91个潜在的候选肺癌分子标记物。基因注解分析显示候选肺癌基因富集了和代谢、TP53通路以及MicroRNA调控等相关的基因。最后,利用Human Protein Atlas数据库及Pubmed对前20候选标记物进行验证, 结果显示大部分候选肺癌基因大多能够得到验证。可见数据挖掘在即将到来的质谱推动的组学大数据时代将发挥重要作用。
关键词:蛋白质组;数据挖掘;肺癌;分子标记物
人类基因组草图的发表迄今已经有15年。在这期间,测序技术的不断成熟以及成本的不断下降促使基因组学在生物医学研究中占据举足轻重的作用[1]。基因微芯片技术以及RNA测序则推动了我们对于基因在RNA水平表达的认识[2, 3]。基于转录组学数据的研究使我们得以发现很多在疾病发生发展中起重要作用的生物标记物,比如肿瘤生物标记。由于技术的限制,我们对于人类蛋白组学的认识一直处于相对落后的状态。 近年来,质谱技术的发展正在催生一个崭新的蛋白质组学时代的到来[4, 5]。蛋白组学数据的存储、分析、解析成为生物信息学的一个新挑战[6]。越来越多的杂志要求研究人员将高通量数据上传到公共数据库,使得普通研究人员借助简单的生物信息学方法能够对这些数据进行挖掘、整合,以自己独特的角度进行数据的再分析。
癌症分子标志物的研究具有重大的临床意义。一方面,分子标记物可以大大提高癌症早期诊断率。目前,前列腺癌特异抗原(PSA)、癌胚抗原(CEA)、及甲型胎儿蛋白(AFP)分别被广泛用于前列腺癌、结肠癌和肝癌等的筛查,为高危人群提供了一个成本较低的筛查手段。另一方面,癌症分子标记物可以为靶向治疗提供新的策略。BCL-ABL融合基因是慢性粒细胞性白血病常见的突变,针对这个突变酪氨酸激酶的靶向药伊马替尼大大提高了慢性粒细胞白血病的生存期。
目前,肺癌在全世界范围内是导致最多癌症死亡的杀手[7]。尽管近年来我们在靶向治疗及早期诊断方面取得了许多激动人心的进展,很多肺癌病人的诊断往往已经到了晚期,使得病人失去手术机会并且对标准化疗方案鲜有反应。对于肺癌分子标志物的界定以及功能学研究有助于我们更深入的认识肺癌发生发展的分子生物学机理,从而能在早期诊断以及靶向治疗方面取得新的突破。
最近,两个研究小组同时发表了人类蛋白组草图[8, 9]。人类蛋白组草图以及更多后续研究将成为一个宝贵的金矿,从而推动我们对于生物标记物的认识。本研究将利用公开发表的人类蛋白质组草图数据[9],结合其他公共数据库的数据,探索潜在的有价值的肺癌分子标记物。
1数据来源与方法
1.1数据来源
原始蛋白组数据来源于http://www.nature.com/nature/journal/v509/n7502/full/nature13319.html
1.2分析方法
R-project开发的开源统计语言R用于进行所有的数据分析以及作图。R编程使用图形界面软件Rstudio。数据录入采用gdata库中的read.xls函数。其他函数均为R基础库中包含的函数。肺癌表达数据来源于5株肺癌细胞系表达的平均值;正常参照蛋白组原始PSM数值进行log10转化。我们定义量化指标以对1 816个肺癌基因表达水平进行界定,即ratio = 肺癌细胞系表达均值/log10(PSM)。
采用马克思普朗克分子遗传研究所开发的在线工具ConsensusPathDB (http://cpdb.molgen.mpg.de/)进行生物通路富集分析。ConsensusPathDB基于KEGG、WikiPathways等数据库。p值大于0.01作为显著性检验的标准。我们使用Panther在线工具(http://pantherdb.org/)进行基于生物功能的基因注解。
1.3论文图文
使用微软Word文本处理软件准备本论文草稿,Inkspace软件准备论文中矢量图制作。
2结果分析
2.1候选肺癌分子标志物筛选
首先,下载5株肺癌细胞系的蛋白质表达数据[9]。这5株肺癌细胞系分别为:A549, h160, H226, H23和p22, 涵盖了常见的肺癌类型。表1为5株肺癌细胞系在ATCC细胞系数据库中的相关信息。在5株肺癌细胞系中检测到蛋白质表达的基因数目高达12 668个。其中,有1 816个基因在5株细胞系中均能检测到蛋白质表达(肺癌共表达基因)。
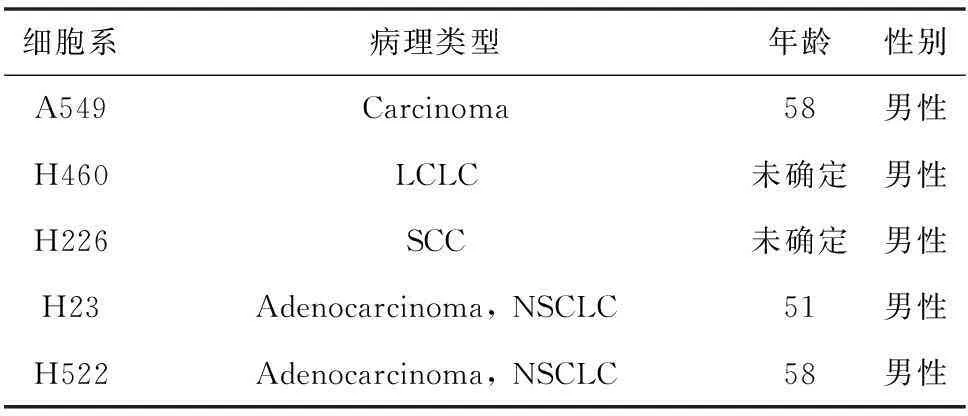
表1 本研究所涉及的肺癌细胞系
接下来,下载涵盖18 097个基因表达的人类蛋白组草图。所有蛋白质表达水平由PSM表示,其中PSM是该蛋白质在ProteomicsDB数据库中的多肽谱配对数。人类蛋白质草图将作为人类蛋白质表达的正常参照,用于和肺癌细胞系蛋白组进行比较从而发现潜在的肿瘤标记物。依次检索1 816个肺癌共表达基因在人类蛋白组草图中的表达水平(PSM),并且进行log10转换,转换后的PSM数值可以同前面下载的肺癌共表达基因进行比较。
为了获得肺癌共表达基因的表达水平,首先将1 816个肺癌共表达基因在5株肺癌细胞中的表达进行平均。然后,采用简化的量化指标以对1 816个肺癌基因表达水平进行界定,即ratio = 肺癌细胞系表达均值/log10(PSM)。这个比率将作为肺癌生物标记物指数。图1(a)为肺癌生物标记物指数的直方图分布,处于柱状图中心位置的基因在肺癌组织的表达水平和在人类蛋白质草图中的表达水平最为接近。而处于柱状图两侧的基因则是表达水平偏离(高于或者低于)人类蛋白组草图表达水平的基因。
利用Ratio对1 816个肺癌共表达基因进行由高到低的排序见图1(b),以筛选出在肺癌组织中表达水平远高于人类蛋白质草图参照的基因。以0.725 8作为log2(ratio)的阈值能够筛选出5%在肺癌中上调表达的基因(合计91个),即肺癌的候选分子标记物。表2为前20的候选分子标记物。

图1 候选肺癌标记物筛选Fig.1 Screening of candidate biomarkers for lung cancer
为了研究哪些生物学功能在肺癌发生发展过程中被富集,基因注解(Gene Ontology)被用于对91个候选肺癌标记物进行分析。 图2只显示了基因注解显著的结果,发现催化活性、蛋白结合等生物学功能被富集,其中包括已知的肺癌相关蛋白NRAS。被富集的生物学功能横跨了细胞表面受体到转录因子及下游基因表达的“信号传递”过程,包括受体活性、 转运蛋白、DNA结合转录因子、 蛋白质结合转录因子、酶调控因子及催化活性。这提示癌症是一个复杂的疾病,细胞内部细胞传递的各个步骤都可能被癌细胞利用产生对癌细胞有利的表型。

图2 候选肺癌生物标记物显著富集的生物功能及其比例Fig. 2 Geneontology analysis for lung cancer biomarkers
为进一步研究这91个肺癌候选标记物所参与的生物通路。采用马克思普朗克分子遗传研究所开发的在线工具ConsensusPathDB获取这91个肺癌候选标记物所富集的生物通路。结果发现三羧酸循环和氧化呼吸链相关基因在肺癌当中被大量富集,支持肺癌在发生发展过程中代谢通路的重塑。值得注意的是,和P53相关的基因在肺癌候选标志物中也被富集,其中包括参与代谢的基因和受P53转录调控的基因。还发现许多肺癌候选标记物受MicroRNA调控,提示基于MicroRNA的药物研发可能为肺癌的治疗提供新的方向,91个候选肺癌生物标记物采用ConsensusPathDB进行功能注解,显示的是显著富集的生物通路。节点大小表示该通路相关基因数目;节点颜色越深,p值越小;节点间连线越粗,两个节点共有基因越多;节点间连线颜色指示候选基因中参与该通路的基因数目,粉色最多,灰色最少 (见图3)。
2.2基于公共数据库及Pubmed的验证
首先,我们采用human protein atlas数据库[10, 11]对前二十个候选肿瘤标记物进行验证。前20个候选标记物中除了PROX1和STMN2外,其他标记物在数据库均有免疫组化数据。我们发现除了YJEFN3、 RAB39A 和LRRC16B 3个基因只有1个病例出现低表达, 大部分候选标记物在肺癌组织中都有低、中、高等不同程度的表达。尽管免疫组织化学的数据和所采用的抗体关系密切,但是大部分候选基因在肺癌的表达能够得到验证(见图4)。

图3 候选肺癌生物标记物富集的生物通路Fig.3 Enrichment of biological pathways for lung cancer biomarkers
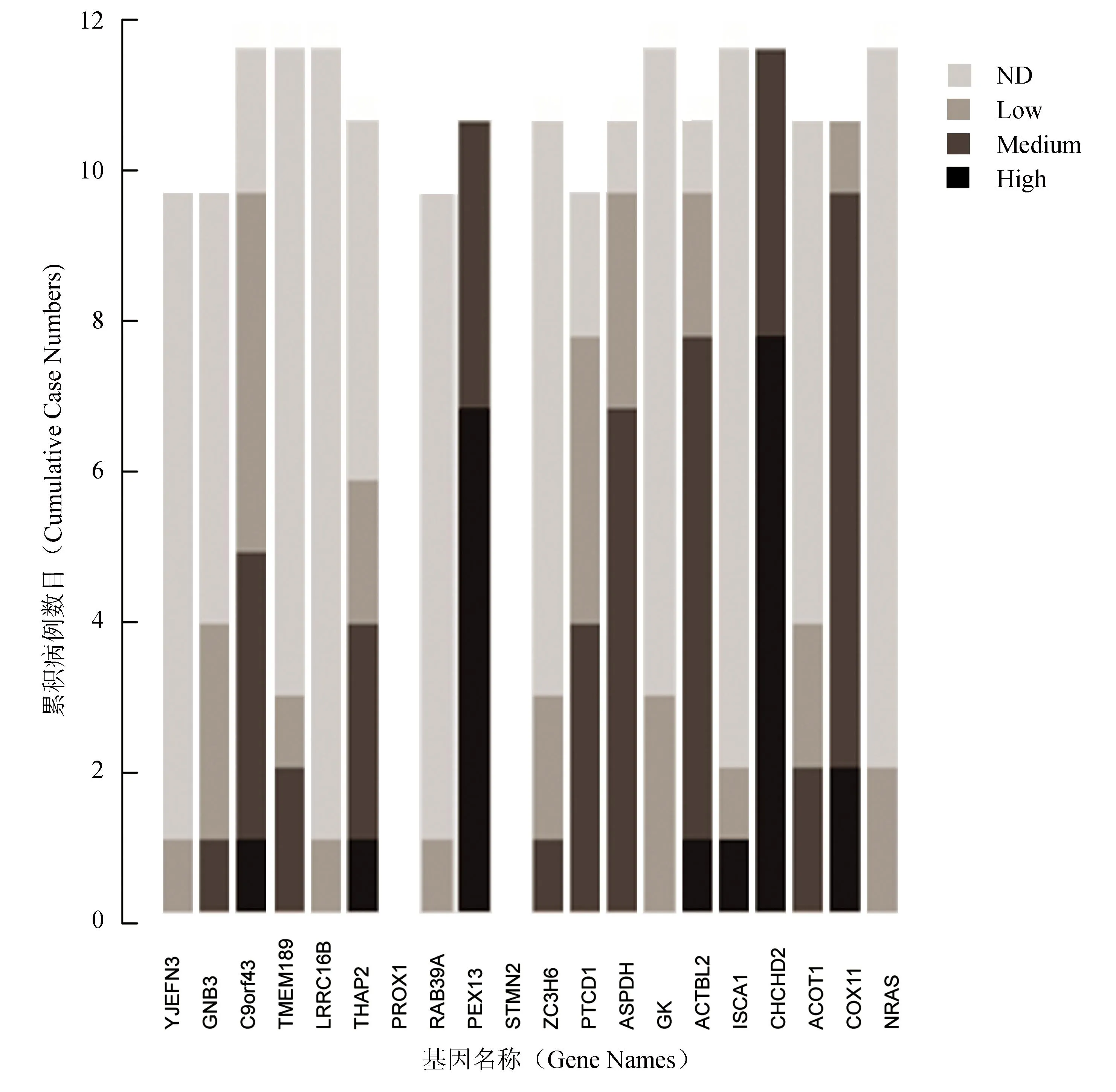
图4 候选生物标记物在人类蛋白质图谱数据库中的验证Fig. 4 Validation of biomarkers in human protein altas
不同数目的肺癌组织用不同抗体对候选生物标记物进行染色,根据染色强度分为高表达 (High),中表达 (Medium),低表达 (Low),无法检测 (ND)。
其次,利用Pubmed对部分候选肺癌基因进行检索。由于对于PROX1和STMN2在人类蛋白质图谱中没有相应的数据可以分析该基因在肺癌组织中的表达,Pubmed将用于进行文献检索。有研究表明PROX1在肺癌中过表达,而采用慢病毒介导的shRNA敲低PROX1则会抑制肺癌细胞的增殖,提示PROX1很可能是一个潜在的肺癌标志物[12]。另外,STMN2作为调控微管动态的基因,是WNT通路的下游。研究表明STMN2在肝癌中高表达并且对于维持肝癌细胞锚定非依赖的生长状态有重要意义[13],而它在肺癌中的作用尚不清楚。
3讨论
利用公共数据库的蛋白质组学数据以及开源统计语言R对已发表的数据进行再分析与再解析,探索肺癌共表达基因作为肺癌分子标志物的可能性。发现NRAS在内的已知肺癌标记物,筛选出一系列具体功能尚未清楚的肺癌候选标记物。进一步的湿实验验证将为最终界定这些基因在肺癌发生发展中的作用以及它们作为肺癌分子标记物的可行性。值得注意的是,该策略也可能富集一些正常肺组织相对一般组织高表达的基因,从而导致一定的假阳性率。比如,PEX13是一个在过氧化物酶体中高表达的基因,其上调有可能是正常肺组织为适应高氧环境的常态,也可能是肿瘤细胞特定代谢进化而来的优势表型[14]。因此,候选标记物具体生物学功能需要进一步实验验证。
基因注解分析显示:筛选出的候选肺癌标志物富集了和代谢、TP53以及MicroRNA调控相关的基因。代谢和TP53通路被富集,说明肺癌发生发展过程中癌细胞进化并且重塑了它们的代谢网络且TP53通路被异常调节。代谢通路的重塑可能和癌细胞的“Warburg”效应相关[15],即癌细胞倾向于上调无氧代谢通路;而P53作为抑癌基因在大多数癌症中有直接突变或者其他相关蛋白的突变,使得P53无法行使正常功能[16]。
随着质谱技术的不断发展,越来越多的实验室开始对蛋白质组学研究产生新的兴趣,这意味着未来会有越来越多的组学数据产生。而大部分组学数据将会被存储在公共数据库如PRIDE, proteomeicsDB等[9, 17]。借助开源软件R对公共数据库中的数据进行二次分析、解析以及整合将有助于获得新的认知。而将这种“干”研究获得的信息用于指导实验设计并进行“湿”实验验证则将成为未来生物医学研究的大趋势[18],即计算生物学与实验生物学的互相补充。过去十几年兴起的系统生物学代表了这一新的趋势,并且已经在生物和医学研究中扮演着重要的角色[19]。
4结论
通过基于公共数据库的数据挖掘筛选出91个潜在的肺癌分子标志物。基因注解分析显示这些肺癌标志物富集了和代谢、TP53网络相关的基因以及MicroRNA靶基因。人类蛋白组草图的发表对于生物医学研究人员有重大意义。蛋白质组学产生的大数据以及这些数据通过公共数据库的共享将深远的影响生物医学研究。
参考文献
[1]MARDIS E R.A decade’s perspective on DNA sequencing technology[J]. Nature, 2011,470(7333):198-203.
[2]SCHENA M, SHALON D, DAVIS R W, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray[J]. Science, 1995, 270(5235):467-470.
[3]WANG Z, GERSTEIN M,AND SNYDER M.RNA-Seq:a revolutionary tool for transcriptomics[J].Nature Reviews Genetics,2009,10(1):57-63.
[4]AEBERSOLD R, MANN M.Mass spectrometry-based proteomics[J]. Nature, 2003,422(6928): 198-207.
[5]NILSSON T , MANN M , AEBERSOLD R, et al.Mass spectrometry in high-throughput proteomics: ready for the big time[J]. Nature Methods, 2010,7(9):681-685.
[6]BOGUSKI M S, MCINTOSH M W. Biomedical informatics for proteomics[J]. Nature, 2003,422(6928):233-237.
[7]HERBST R S , HEYMACH J V , LIPPMAN S M. Lung cancer[J]. New England Journal of Medicine, 2008,359(13):1367-1380.
[8]KIM M S , PINTO S M , GETNET D , et al.A draft map of the human proteome[J]. Nature, 2014,509(7502): 575-581.
[9]WILHELM M , SCHLEGL J , HAHNE H, et al. Mass-spectrometry-based draft of the human proteome[J]. Nature, 2014,509(7502):582-587.
[10]UHLEN M , OKSVOLD P , FAGERBERG L, et al.Towards a knowledge-based Human Protein Atlas[J]. Nature Biotechnology, 2010, 28(12):1248-1250.
[11]UHLéN M, FAGERBERG L, HALLSTRöM BM, et al. Proteomics. Tissue-based map of the human proteome[J]. Science, 2015,347(6220):1260419.
[12]ZHU S H , SHAN C J , WU Z F, et al.Proliferation of small cell lung cancer cell line reduced by knocking-down PROX1 via shRNA in lentivirus[J]. Anticancer Research, 2013, 33(8): 3169-3175.
[13]LEE H S , LEE D C , PARK M H , et al. STMN2 is a novel target of beta-catenin/TCF-mediated transcription in human hepatoma cells[J]. Biochemical ang Biophysical Research Communications, 2006,345(3):1059-1067.
[14]ORUQAJ G , KARNATI S , VIJAYAN V , et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-beta signaling[J]. Proceedings of the National Academy of Sciences, 2015,112(16):E2048-2057.
[15]KOPPENOL W H , BOUNDS P L, DANG C V.Otto Warburg's contributions to current concepts of cancer metabolism[J]. Nature Reviews Cancer, 2011, 11(5):325-337.
[16]BIEGING K T, MELLO S S , ATTARDI L D. Unravelling mechanisms of p53-mediated tumour suppression[J]. Nature Reviews Cancer, 2014,14(5):359-370.
[17]VIZCAINO J A , COTE R G , CSORDAS A, et al.The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013[J]. Nucleic Acids Research, 2013,41(Database issue):D1063-1069.
[18]黄晓韵,曹波,杨跃.基于SAS的多元统计方法实现芯片数据挖掘[J].生物信息学,2010,8(2):147-149.
HUANG Xiaoyun, CAO Bo ,YANG Yue.Microarray data mining is achieved by multivariate statistics based on SAS[J]. Chinese Journal of Bioinformatics,2010,8(2):147-149.
[19] HOOD L. Systems biology and p4 medicine: past, present, and future[J]. Rambam Maimonides Medical Journal,2013,4(2): e0012.
Pilot study on biomarkers for lung cancer based on the draft of human proteome
ZHU Yanyan1,2, LUO Zhifen1,2,CUI Yongxia1,2, LU Chuangxin1,2, ZHOU Yun1,2*
(1.OncologyUnit,People’sHospital,Zhengzhou450003,China;2.OncologyUnit,People’sHospital,ZhengzhouUniversity,Zhengzhou450003,China)
Abstract:Traditional exploration over lung cancer molecular marker has been relied on genome or transcriptome research, while exploration at the protein level has been limited by throughput. Mass spectrometry based proteome research has started to generate global proteome data for normal and cancer tissue. Using the open source statistical language R, we mined the publicly available data of human proteome draft and lung cancer proteome for screening for candidate molecular markers for lung cancer.We identified 91 candidate biomarkers for lung cancer.Gene ontology analysis suggested that candidate lung cancer biomarkers have enriched genes associated with metabolism, TP53 network and microRNA regulation. Top hits on the list were then validated with Human Protein Atlas database and Pubmed, which shows that most hits can be validated. We believe data mining has an important role to play in the big omic data era that is being ushered in by mass spectrometry.
Keywords:Proteome;Data mining;Lung cancer;Biomarkers
中图分类号:Q51;Q279
文献标志码:A
文章编号:1672-5565(2016)01-043-06
doi:10.3969/j.issn.1672-5565.2016.01.08
作者简介:朱琰琰,女,研究方向:肿瘤学、系统医学;E-mail:xjtu100@163.com.*通信作者:周云,男,主任医师,研究方向:肿瘤学;E-mail:zlk2092@126.com.
收稿日期:2015-10-26;修回日期:2015-12-16.
猜你喜欢
保健医苑(2023年2期)2023-03-15
中国临床医学影像杂志(2022年2期)2022-05-25
大众投资指南(2021年35期)2021-02-16
电力与能源(2017年6期)2017-05-14
中国中医药信息杂志(2016年7期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
信息通信技术(2015年6期)2015-12-26
医学研究杂志(2015年12期)2015-06-10
郑州大学学报(医学版)(2015年1期)2015-02-27
河南科技(2014年23期)2014-02-27
