
四种常用的生物序列比对软件比较
2016-04-26 09:29:05陈凤珍操利超严志祥
生物信息学 2016年1期
陈凤珍,李 玲, 操利超, 严志祥
( 深圳华大基因研究院, 深圳 518083)
四种常用的生物序列比对软件比较
陈凤珍,李玲, 操利超, 严志祥*
( 深圳华大基因研究院, 深圳 518083)
摘要:随着高通量测序技术的快速发展,下一代测序技术也迅速发展为生物领域中的主流技术,而理解下一代测序数据最重要的一步是比对。比对是进行后续生物信息分析的基石,也因此催生了很多比对软件。本文主要选取了四种常用的比对软件Bowtie2、BWA、MAQ和SOAP2,对这四种软件及算法进行综述,并通过实际测序数据对四种软件进行比较和评估,为生物学研究者选择最佳的短序列比对软件提供理论和实践依据。
关键词:下一代高通量测序; 比对软件; 生物信息
1引言
随着新一代测序(Next-generation sequencing,NGS)的蓬勃发展,核酸测序成本已大大降低,高通量测序方法已被广泛应用到DNA测序[1]、RNA测序[2]、表观遗传测序[3-4]等研究。然而,无论使用何种生物测序技术和研究方法,理解这些数据的最重要的一步是序列比对分析。序列比对是将已有基因组序列作为参考基因序列(Reference),将短序列与参考基因序列进行序列比对, 并在参考基因序列上进行精确定位。通过序列比对可以发现生物序列中的功能、结构和进化的信息。目前已有上百种序列比对工具,面对如此多的比对工具,很多生物信息分析人员通常自由的选择比对工具,而没有考虑到比对工具的特点,准确性等。然而,不同的比对软件,对同一个数据集都有可能得出大相径庭的结果[5];同一算法设置不同的参数,其结果也相差很巨大。如果选择了一个不合适的工具,将导致结果偏差甚至是错误,可能得到错误的研究结论。因而选择合适的比对工具,对于生物研究而言显得特别重要。
在Nuno A. Fonseca等人[6]的对60多种比对软件比较统计分析中,发现Bowtie2[7]、BWA[8]、MAQ[9]和SOAP2[10]被引用的次数相对其他几十种软件较多,其中Bowtie2引用率(Citations/Years)为363.42,BWA为224.20,MAQ为251.66,而SOAP2为99.38,SOAP2的前版本SOAP为104.41。因而在本研究中,主要选取了这四种常见的比对工具进行评估比较。根据比较结果分析,Bowtie2、BWA和SOAP2处理高通量短序列数据比对问题时,计算速度快,内存使用量低,具有高效的实用性;在同等条件下,MAQ的运行速度较慢。 Bowtie2、BWA的比对率相比于SOAP2和MAQ高。BWA软件与Bowtie2软件比对的重复率较高,MAQ较低。
2四种比对软件及算法
2.1四种比对软件介绍
Bowtie2是一个超高速的,节约内存且灵活与成熟的短序列比对软件,比较适合下一代测序技术。通常使用全文分索引(FM-index)以及Burrows-Wheeler 变换(BWT)索引基因组使得比对非常快速且内存高效,但是这种方法不适合于找到较长的、带缺口的序列比对。
BWA主要应用二代测序后的大量短小片段与参考基因组之间的定位比对。需要先对参考序列建建立索引,BWA也是基于 BWT和 FM-Index 理论来对参考基因组做索引。根据测序方法的不同,有单末端序列(Single-end,SE)比对和双末端序列(Pair-end,PE)比对。
MAQ是使用质量分数推导序列和比对序列的一致性的短序列比对工具,并且MAQ充分利用配对信息,估计每个比对read的错误的概率,同时也使用贝叶斯统计模型来评估最后的基因型错误概率。
SOAP2是短寡核苷酸比对程序(Short Oligonucleotide Alignment Program)的一个显著改进版本,它减少了计算机内存使用,并极大地提高了比对速度。SOAP2使用一个Burrows Wheeler Transformation(BWT)压缩索引替代种子策略在主存储器中索引参考序列。SOAP2适合于单末端片段和双末端片段。此外,该工具也支持多种文本和压缩文件格式。
2.2四种比对软件算法
对于成千上万条的短序列的比对分析,目前,大多数算法是通过建立索引来加快比对的速度。常用的数据结构有哈希表法和基于BWT (Burrows-wheeler transform) 的后缀树两种。
哈希表法的算法核心思想是采用种子序列定位及延伸算法(Seed-and-extend algorithm)[11],通过扫描参考基因组序列,对参考基因组序列建立哈希表,将序列分成一定长度的小片段,这种小片段也被称之为种子。然后,在目标序列中查找和种子序列相同的片段并标记,以这些标记点为锚点向左右按一定规律延伸比对,将不合条件的舍弃,符合条件的结果将输出保存。采用基于哈希表数据结构的比对算法的软件包括MAQ。
罗楼组以泥岩为主,兼有火山碎屑岩,可分下段和上段。下段主要由泥岩组成。底部夹少量泥质灰岩,下部夹有一层厚约1 m的玻屑凝灰岩,中、上部夹凝灰质泥岩,泥岩呈灰-深灰色,薄层状,主要由水云母(95%~98%)组成,含少量石英(1%~5%)和微量铁质和有机质;下部泥岩时见生物潜穴遗迹。
后缀树法是一种n叉树,n为字母表大小。每个节点表示从根节点到此节点所经过的所有字符组成的字符串,它的根结点不包含任何信息,是一种以牺牲存储空间来降低序列查询时间的字符串预处理方式。为了提高空间利用率,Ferragina 和Manzini 提出了FM (Full-text minute-space)-index 算法,FM是一种基于BWT (Burrows-wheeler transform)的全文本压缩索引结构, BWT算法是通过统计基因组序列中各个碱基出现次数,将相同碱基尽量排列在一起,压缩基因组序列的索引数据结构,将基因组序列的索引数据结构重排列,实现短序列在基因组中候选位点的快速搜索,减少内存占用率。例如人类基因组约3GB,若不使用FM-index将要用12GB内存存储,超过了计算机内存使用限度,而如果使用FM-index, 每隔数行建立一个索引,人类基因组占用的内存可缩小到约1.3GB,这样普通的计算机就可以进行分析。采用BWT转换的软件有Bowtie2和SOAP2,BWA。
虽然Bowtie2、SOAP2和BWA都采用了BWT算法,然而三种软件还有差别。其中Bowtie2采用Ferragina 和Manzini 提出的FM (Full-text minute-space)-index算法,为基因组序列创建具有后缀矩阵特性的 FM 索引数据结构,实现短序列的快速搜索;SOAP2则采用的是 BWT 算法压缩基因组序列哈希表索引数据结构进行精确匹配,采用“分割短序列策略”(Split-read strategy)进行不精确匹配,比对速度显著提高且内存使用量显著地降低。最后,BWA 软件是采用 BWT 算法压缩来构建基因组序列前缀树(Prefix tree)数据结构,通过对压缩数据结构自顶向下遍历进行反向搜索,其比对计算过程中内存覆盖区域相对较小,计算时间并不随基因组的大小而变化。
基于哈希表法和基于BWT的后缀数法数据结构的算法都有利于提高比对效率,区别在于哈希表法占用的内存空间大,产生的种子匹配多,然而哈希表法具有较高的匹配敏感性和准确性。有利于发现SNPs和突变。可用于局部匹配或从大量数据中搜索匹配点以及跨物种序列间的比对。而后缀树法可以有效减少不精确匹配,并可避免比对过程中做无用功,这个特点适用于相同物种之间相似性高的序列比对和寻找保守区。
2.3四种比对软件比较
选择合适的软件要根据软件适用的数据类型,适宜测序平台,数据格式,适宜的reads长度等进行全面考虑,做出选择。表1中对四种比对软件分析的序列类型,可用于分析的测序平台,输入和输出数据格式,最小和最大reads长度及软件是否开源进行了详细的分析和比较。从表中可以看出在适宜测序平台方面,SOAP2就受到限制,只适用于Illumina平台,BWA适用的平台最广。在适宜的reads长度方面,BWA、MAQ适用的范围较窄。最后,根据软件的输入输出格式,MAQ的适用范围更广。
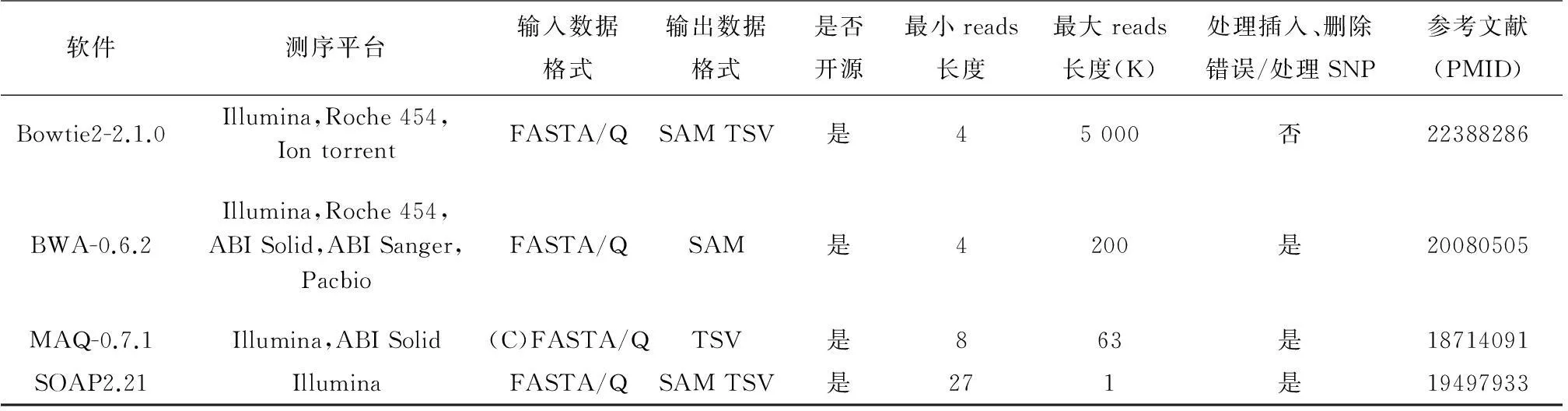
表1 四种比对软件比较
3软件评估实验
3.1实验数据
3.2软件运行环境
32G内存,16核处理器,linux操作系统服务器。
3.3结果评估
四种软件的比对率和时间消耗如表3。从表3可以看出BWA和Bowtie2的比对率较高,而SOAP2的时间更高效,MAQ相对来说较慢。

表2 实验数据
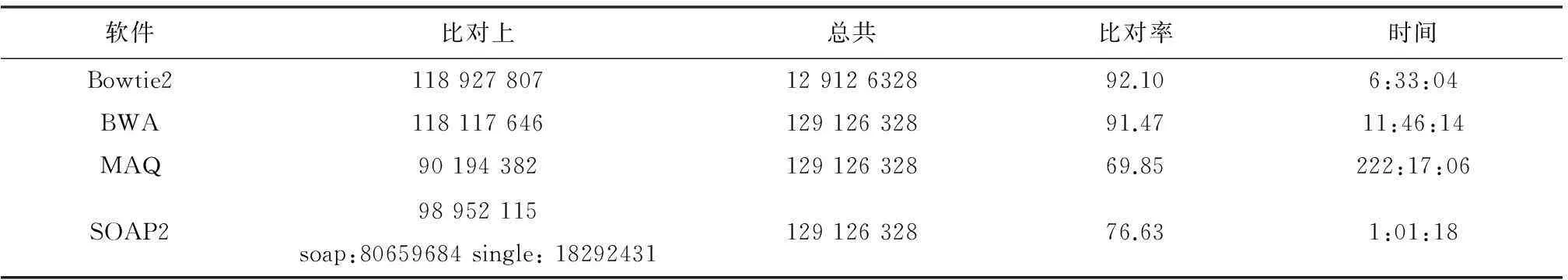
表3 四种比对软件比对率和比对时间
从四种软件比对的reads重复数两两比较可以看出,Bowtie2和BWA比对上的reads重复数较高,Maq和其他三种软件比对上的reads重复数较低,如图1。将四种软件同时比较时,发现BWA比对软件和其他三种软件不重复的reads数最少,只有62 134条,Bowtie2和其他三种软件不重复的reads数最多,为466 792条,如图2。
从实验结果看出Bowtie2和BWA的比对率相比于SOAP2和MAQ高。BWA软件与Bowtie2软件比对的重复率较高,MAQ较低,可能与选取的实验数据相关,本实验选取的是高覆盖度的HPV全基因组测序数据,BWA比对工具比较适合全基因组测序数据的比对分析。
4讨论
通过比较和实验研究发现,Bowtie2、BWA、MAQ和SOAP2四种软件在处理高通量短序列数据比对问题时,计算速度较快,内存使用量较低,具有高效的实用性。 但是,这四种常用的分析软件都只对短序列分析较为适合,然而,第三代测序技术正在快速的发展,必将成为未来的主流技术。第三代测序技术相比于第二代测序技术特点之一是读长长。因而开发高准确性的适合第三代测序数据的长序列比对工具是未来研究的主题。
对于比对分析一个常见的问题是,哪一个分析工具是本研究最适合的。一个最好最适合的比对工具不光要考虑数据的类型,一个重要的方面包含比对工具是否和比对下游的分析和分析工具结合紧密,更包含比对的工具的速度和准确性。但是目前,评估一个比对工具的准确性和速度仍然很难,主要的困难是缺乏不同测序技术和研究方法的金标准数据集,因为不同的比对软件,不同的数据集,数据类型,数据大小等都有可能导致比对准确度和时间偏差。因而创建适合的金标准数据集对于比对工具的评估和研究特别重要。

图1 四种比对软件比对上的读长两两比较Fig. 1 The multiple compration of mapped reads using the selected software
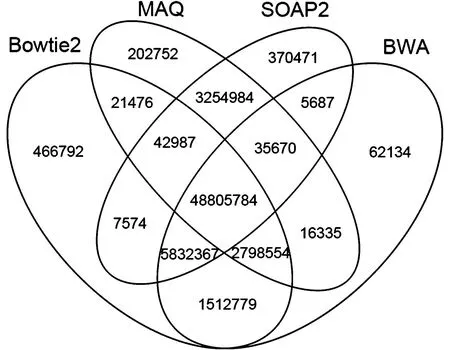
图2 四种软件比对上的读长比较Fig. 2 The compration of mapped reads using the selected software
5结论
对二代测序的四种常用比对软件的算法进行了总结,并对四种软件的适用性和性能等方面进行了对比,同时利用实际的基因组数据进行测试分析,归纳总结,给出软件选择的参考建议,为研究人员选择适合的比对分析工具提供参考。
参考文献
[1]MARDIS E R. Next-generation DNA sequencing methods[J]. Annual Review of Genomics and Human Genetics,2008, 9: 387-402.
[2]WANG ZHONG, GERSTEIN M, SNYDER M. RNA-Seq: a revolutionary tool for transcriptomics[J]. Nature Reviews Genetics, 2009, 10:57-63.
[3]PARK P J. ChIP-seq: advantages and challenges of a maturing technology[J]. Nature Reviews Genetics,2009, 10(10): 669-680.
[4]MEISSNER A, MIKKELSEN T S, GU H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells[J]. Nature,2008, 454(7205):766-770.
[5]NEKRUTENKO A, TAYLOR J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility[J]. Nature Reviews Genetics, 2012, 13(9):667-672.
[6]FONSECA N A, RUNG J, BRAZMA A, et al. Tools for mapping high-throughput sequencing data[J]. Bioinformatics,2012, 28(24):3169-3177.
[7]LANGMEAD B, SALZBERG S L. Fast gapped-read alignment with Bowtie 2[J]. Nature Methods,2012, 9(4):357-359.
[8]LI HENG, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics,2009, 25(14):1754-1760.
[9]LI HENG, RUAN JUE, DURBIN R. Mapping short DNA sequencing reads and calling variants using mapping quality scores[J]. Genome Research,2008, 18(11):1851-1858.
[10]LI Ruiqiang, YU Chang, LI Yingrui, et al. SOAP2: an improved ultrafast tool for short read alignment[J]. Bioinformatics,2009, 25(15):1966-1967.
[11]LI Heng, HOMER N. A survey of sequence alignment algorithms for next-generation sequencing[J]. Briefings in Bioinformatics,2010, 11(5):473-483.
Comparison of four common biological sequence alignment tools
CHEN Fengzhen, LI Ling, CAO Lichao,YAN Zhixiang*
(BGI-Shenzhen,Shenzhen518083,China)
Abstract:With the rapid development of high-throughput sequencing technology,Next-generation sequencing technology has rapidly developed into a mainstream technology in the biological field. Alignment is the key step in understanding the sequence data and also it is the cornerstone for bioinformatics analysis. And thus gave birth to a lot of alignment tools. In this paper, four common biological sequence alignment tools Bowtie2, BWA, MAQ and SOAP2 were selected to evaluate and compare using the whole genome sequencing data of HPV. And a comparison of four tools from many perspectives such as algorithm and suitable sequencing platforms was given. Hopefully the research can provide theoretical and practical basis for researchers to select the best biological sequence alignment tools.
Keywords:Next generation sequencing; Alignment tools; Bioinformatics
中图分类号:Q-31
文献标志码:A
文章编号:1672-5565(2016)01-056-05
doi:10.3969/j.issn.1672-5565.2016.01.10
作者简介:陈凤珍,女,生物信息工程师;E-mail:chenfengzhen@genomics.cn.*通信作者:严志祥,男,博士,生物信息高级工程师;E-mail:yanzhixiang@genomics.cn.
基金项目:国家自然科学基金资助项目(U1301252)。
收稿日期:2016-01-19;修回日期:2016-03-08.
