
两例先天性肾上腺发育不良伴低促性腺激素性性腺功能减退症
2016-04-19 03:19袁进磊张会娟刘艳霞刘艳杰
河南医学研究 2016年2期
袁进磊 张会娟 刘艳霞 刘艳杰
(郑州大学第一附属医院 内分泌科 河南 郑州 450052)
两例先天性肾上腺发育不良伴低促性腺激素性性腺功能减退症
袁进磊张会娟刘艳霞刘艳杰
(郑州大学第一附属医院 内分泌科河南 郑州450052)
【摘要】目的通过探讨两例先天性肾上腺发育不良(adrenal hypoplasia congenital,AHC)伴低促性腺激素性性腺功能减退(hypogonadotropic hypogonadism,HHG)患者的临床表现,增强对先天性肾上腺发育不良症的认识,为今后更及时有效地诊治肾上腺疾病提供帮助。 方法收集患者的临床资料、生化检查、影像学检查结果及家族史,分析其发病原因。 结果患者临床表现、实验室检查和影像学检查符合AHC诊断。患者糖皮质激素替代治疗后,症状改善。结论AHC患者可伴有低促性腺激素性性腺功能减退症,包括性发育不良和假性性早熟。对于临床上存在肾上腺皮质功能减退、HHG患者,应该考虑到该疾病可能。
【关键词】肾上腺皮质功能减退;低促性腺激素性性腺功能减退症;假性性早熟
先天性肾上腺发育不良(adrenal hypoplasia congenital,AHC)有时是致命的,可以由肾上腺发育不良或缺乏引起。这种情况最先在1948年被病理学家Sikl报道:一个有“皮肤色素沉着”的男孩,刚出生没几周就死于失盐性肾上腺危象。AHC常常伴有低促性腺激素性性腺功能减退症或假性性早熟。AHC为X-连锁的隐性遗传性疾病,女性杂合子为携带者,男性患病。本文将探讨两例AHC患者,他们为同胞兄弟,均表现出肾上腺功能减退症状,但哥哥表现出性发育不良症状,弟弟出现性早熟。对哥哥用糖皮质激素替代治疗后症状缓解。AHC是一种罕见疾病,本文对AHC病例的探讨有助于进一步认识和了解这种疾病,提高对该病的诊疗水平。
1资料与方法
1.1病例资料两例疑有先天性肾上腺发育不良的患者,其为同胞兄弟,经过体格检查和实验室检查、影像检查,确诊为AHC。
患者1,男22岁。以“全身皮肤发黑22 a,加重15 a,双膝关节疼痛1周”为主诉于2014年12月12日入院。伴发热、食欲减退、双下肢无力,测尿17酮类固醇(17-ketosteroide, 17-KS)分别为19.6 μmol/d、27.6 μmol/d,血睾酮1.8 ng/dl,醛固酮、电解质无异常,诊断为“①肾上腺皮质增生症;②上呼吸道感染”。给予“强的松口服,2.5 mg/次,2次/d”治疗,未按医嘱服药。至北京某医院就诊,诊断为“Addison氏病”,给予“醋酸氢化可的松(20 mg)早、晚各10 mg口服”,食欲减退、双下肢无力缓解。2 a前自行停用“醋酸氢化可的松”,未出现食欲减退。2014年12月2日食凉皮、喝饮料后出现呕吐、腹泻,至当地医院,给予“醋酸泼尼松针”治疗,2014年12月5日呕吐、腹泻、腹痛缓解,出现双膝关节疼痛,伴双膝关节活动受限,以“①Addison氏病;②膝关节活动受限待查”收入院。患者系第1胎第1产,出生时体质量约3.6 kg。父母非近亲结婚,均体健,1弟患“先天性肾上腺发育不良”,其他家族成员未发现患有相关疾病。患者无业,无职业病史,无毒物、污染物接触史,无药物、食物过敏史。体格检查:体温36.8 ℃,脉搏58次/min,呼吸18次/min,血压110/80 mm Hg,身高184 cm,体质量68 kg,BMI 20.01 kg/m2,体型偏瘦,全身皮肤发黑,以双侧面颊、口唇、牙龈黏膜、双手第2指关节、双足趾关节显著,其中双足第2~4趾趾关节伸侧可见散在白斑,唇上无胡须,喉结缺如,腋毛缺如,阴毛发育呈Tanner分期2期,双侧睾丸体积约5 ml,发育呈Tanner分期2期,质软,阴茎牵拉长7.0 cm,发育呈Tanner分期2期(见图1)。双膝关节活动受限,可伸至约90°,左踝关节畸形。
实验室检查:性激素6项:促卵泡刺激素(follicle-stimulating hormone,FSH)、促黄体生成素(luteotropic hormone,LH)、雌二醇(estradiol,E2)、孕酮(progesterone,P)、睾酮(testosterone,T)、泌乳素(prolactin,PRL)、17α羟孕酮(17α-Hydroxyprogesterone,17OHP)、游离三碘甲状腺原氨酸(free triiodothyronine,FT3)、游离甲状腺素(free thyroxine,FT4)、促甲状腺素(thyroid stimulating hormone,TSH)(见表1);促性腺激素释放激素(gonadotrpin-releasing hormone,GnRH)激发试验示低平曲线(见表2、图3);血皮质醇偏低,促肾上腺皮质激素(adreno-cortico-tropic-hormone,ACTH)偏高(见表3);骨标志物:总Ⅰ型胶原氨基酸端延313.6 ng/ml,维生素D313.6 ng/ml,骨钙素32.2 ng/ml。β胶原特殊序列测定2.3 ng/ml;血常规:白细胞数17.5×109/L,红细胞数3.60×1012/L,血红蛋白105.0 g/L,血小板总数476×109/L,中性粒细胞14.1×109/L。凝血功能:D-二聚体1.16 μg/ml,凝血酶原时间8.00 S,凝血酶原时间活动度138.10%,国际标准化比率0.72。血脂:甘油三酯1.88 mmol/L。血沉132 mm/h。尿粪常规、电解质、肝肾功、传染病4项无异常。C反应蛋白、风湿全套、类风湿全套、心肌酶、超敏肌钙蛋白、生长激素、24 h尿醛固酮无异常。
影像检查:心电图示:窦性心动过缓;MRI:垂体MRI平扫未见明显异常,右侧上颌窦囊肿;DR示:左右膝关节未见明显异常;彩超示:心内结构及功能未见明显异常,甲状腺弥漫性回声改变伴血流稍增多,双侧睾丸体积小,左侧精索静脉曲张;CT示:双侧肾上腺萎缩(见图5)。
患者2,为患者1弟弟,12岁,患儿10个月时家属发现阴茎发育,能勃起(具体不详),查ACTH 158.0 pg/ml,皮质醇(COR)(16点)13.1 μg/dl,诊断为“性早熟?”,至北京某医院就诊,查阴茎长6 cm,周径6.5 cm,睾丸 4~5 ml,阴阜有色素沉着,阴毛Ⅰ期,手腕DR示骨龄3岁,诊断为“先天性肾上腺皮质增生症-21羟化酶缺乏”,给予“氢化可的松(20 mg)早、晚各10 mg口服”治疗。2 a前出现喉结突出、变声、胡须生长。现阴茎长10 cm,睾丸9 ml。父母非近亲结婚,均体健,1兄患“先天性肾上腺发育不良”,其他家族成员未发现患有相关疾病。患者为学生,无职业病史,无毒物、污染物接触史,无药物、食物过敏史。体格检查:身高162.5 cm,体质量59 kg,BMI 22.34 kg/m2,牙龈存在色素沉着,阴茎牵拉长10 cm,睾丸12 ml(见图2)。
实验室检查:性激素6项(见表1);GnRH激发试验示低平曲线(见表2、图4);
24 h游离皮质醇(见表3);(立位)肾素活性0.01 ng/(ml·h),血管紧张素Ⅱ67.834 pg/ml,醛固酮35.751 pg/ml;血常规、心肌酶、电解质、肝肾功、血脂无明显异常。

影像检查:DR示:左右膝关节未见明显异常;彩超示:双侧睾丸等大,左侧约38 mm×19 mm×12 mm,左侧精索静脉曲张;CT:右侧肾上腺未见显示,左侧肾上腺较小,肝脏点状钙化(见图6)。

图1 患者1阴茎分期为Tanner 2期 图2 患者2的阴茎大于同龄人,Tanner 3期

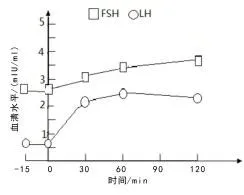
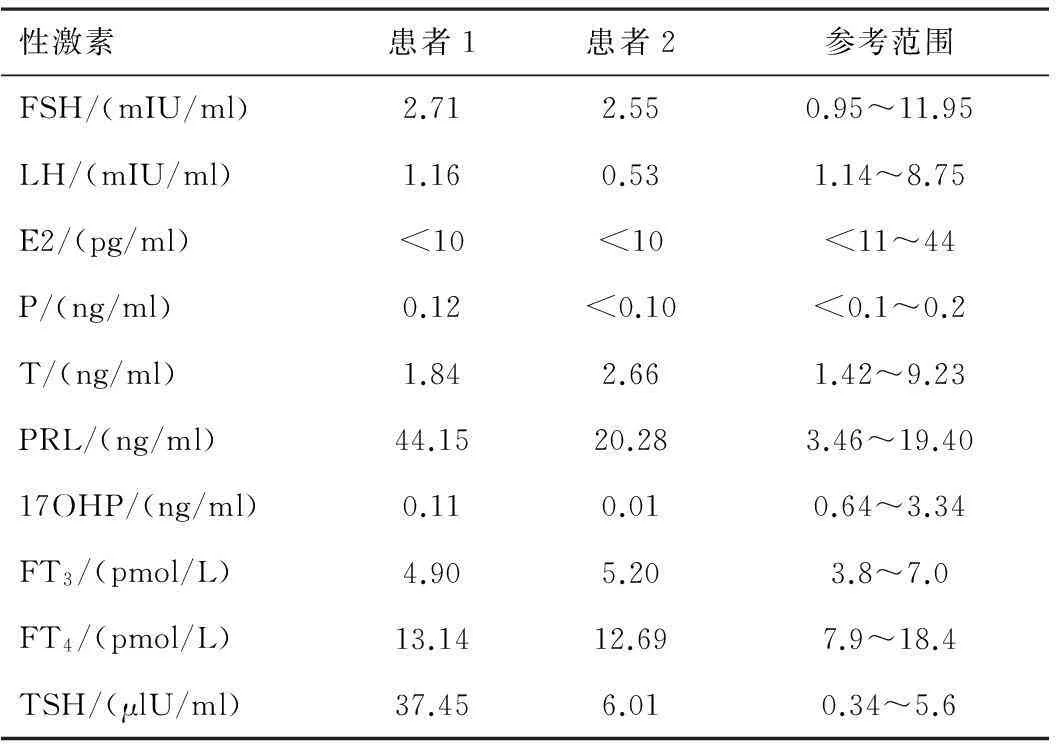
图3 患者1GnRH激发试验曲线 图4 患者2GnRH激发试验曲线
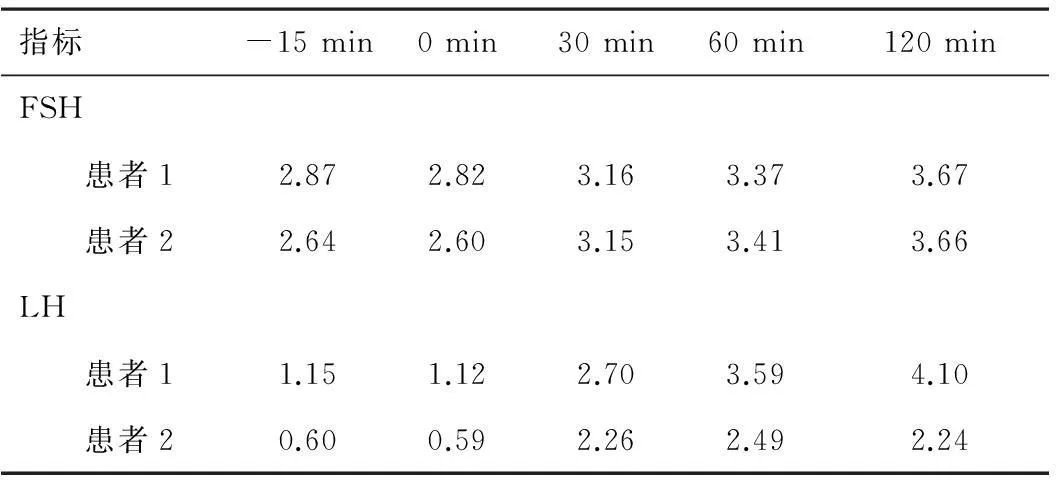
表2 两例患者GnRH兴奋试验(mIU/ml)

表3 两例患者ACTH和皮质醇节律
1.2治疗方法患者1于入院的第1、2日内迅速补充生理盐水2 000~3 000 ml/d并补充葡萄糖液以避免低血糖。给予“强的松(5 mg)早5 mg、晚2.5 mg口服”激素替代治疗,逐渐减量,2 d后患者恶心、呕吐、双膝关节疼痛、双膝关节活动受限缓解。患者2拒绝治疗,后失访。
2结果
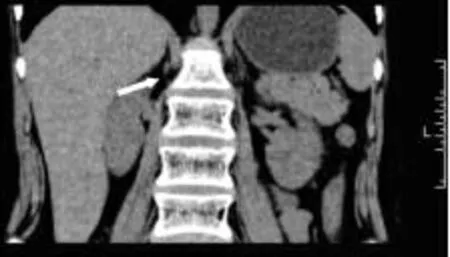
病例1患者儿童期起病,表现有典型的肾上腺皮质功能不全症状,高ACTH、低皮质醇血症,肾上腺CT示双侧肾上腺萎缩,临床高度疑诊AHC。病例2患者实验室检查示高ACTH、低皮质醇血症,CT示右侧肾上腺未见显示,左侧肾上腺较小,考虑诊断AHC。
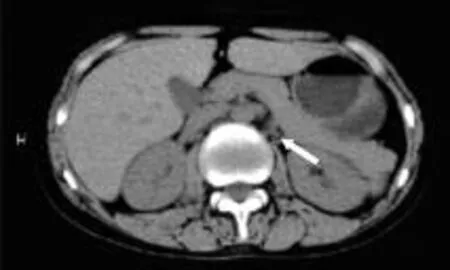
图5 患者1双侧肾上腺萎缩
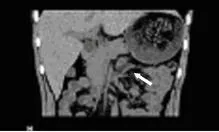
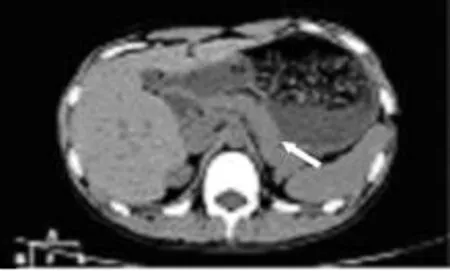
图6 患者2右侧肾上腺未显示,左侧肾上腺较小
3讨论
AHC是一种少见的遗传性疾病,典型的表现是男性患儿由于盐皮质激素(醛固酮)的缺乏,以及糖皮质激素(可的松/皮质醇)的缺乏表现出失盐危象,如纳差、呕吐、腹痛以及低体质量、喂养困难、营养状况差、血管闭塞、不明原因的突然死亡[1]。大部分患者在表现出生殖功能异常之前,就已经表现出肾上腺功能减退以及青春期发育延迟的特点。很大比例的患者青春期性发育延迟,生育能力受损,这是由于低促性腺激素和精子形成的原发性缺陷所致[2]。另有少部分患者表现为外周性性早熟,依赖ACTH的性早熟及中枢性性早熟,表现为阴毛增多、阴茎增长、睾丸体积增大和骨龄提前。性早熟的机制不清楚,可能是多因素所致,包括睾丸间质细胞“自治”,胎儿肾上腺持续产生雄激素,类固醇生成细胞产生的ACTH对睾丸的持续刺激,性腺类固醇对下丘脑-垂体-性腺轴的负反馈减弱。
病例1患者现年22岁,出生时家属发现其全身皮肤发黑,长期仅诊断为Addison氏病。根据其肾上腺皮质功能不全及性腺功能减退症状,血ACTH升高,皮质醇低,GnRH兴奋试验FSH和LH呈低平状态,影像学检查肾上腺萎缩,结合患者未合并甲状旁腺功能减退、1型糖尿病等其他内分泌疾病,可与自身免疫性多内分泌腺瘤综合征(APS)及其他引起慢性肾上腺皮质功能减退的疾病如先天性肾上腺增生等鉴别[3]。
大多数AHC患者到青春期表现出HHG而无青春发育、青春晚发育或发育不完全,是该病的特征之一。这提示在HHG患者的鉴别诊断中应该注意肾上腺皮质功能的评估,以尽早发现这些患者。
病例2患者临床高度疑诊AHC,与其兄诊断相同,但其睾丸体积较大,表现为假性性早熟,曾误诊为21羟化酶缺乏。患者首先出现的临床表现是肾上腺皮质功能减退,在儿童期诊断AHC应与发病率更高的先天性肾上腺增生中的21羟化酶缺乏鉴别,两者都可以表现为肾上腺皮质功能低减,ACTH升高,但两者的不同是21羟化酶缺乏患者血17-OHP和血睾酮水平偏高合并肾上腺皮质增生,与患者不符。推测临床过高的ACTH水平会刺激睾丸间质细胞,导致该男孩出现非促激素依赖青春期前性早熟。
有低促性腺激素性性腺功能减退的患者应考虑本病,可通过基因测定进一步确诊。对于低促性腺激素性性腺功能减退,睾酮替代疗法能够刺激男性化,替代治疗的剂量为250 mg/d。人类慢性促性腺激素也被用于刺激睾酮的产生。为了促使睾丸发育,使其分泌雄激素和生成精子两个功能都得以获得,应首先考虑用GnRH脉冲治疗或人绒毛膜促性腺激素(human chorionic gonadotropin,hCG)+人绝经期促性腺激素(human menopausal gonadotropin,HMG)间断肌内注射治疗[4]。肌内注射hCG+HMG,能促使睾丸容积增大,性激素水平升高,精子生成,第二性征发育。此外,曾有AHC患者通过辅助生殖技术应用睾丸精子提取物进行胞浆内注射(testis sperm extraction and intracytoplasmic injection,TESE-ICSI),最后成功生育后代。尚有患者用睾酮庚酸盐治疗注射,6 a后睾丸激素替代治疗被促性腺激素治疗(FSH 150 UI和LH 150 UI每周3次,以及hCG 1 500 U每周2次)代替。但是治疗20个月后未达到预期的效果,即患者的睾丸激素水平正常,睾丸尺寸正常,但精子缺乏仍然未得到改善[5]。
通过上述两例病例,能够增进对先天性肾上腺发育不良伴低促性腺激素性性腺功能减退症的了解和认识,引起医学工作者对该病的重视,提高该病的确诊率,为尽早地发现与治疗这种疾病提供帮助。
参考文献
[1]Unmesh J,Rebecca M,Harris,et al.Hypogonadotropic hypogonadism in subjects with DAX1 mutations[J].Mol Cell Endocrinol,2011,346(1-2):65-73.
[2]Frapsauce C,Ravel C,Legendre M,et al.Birth after TESE-ICSI in a man with hypogonadotropic hypogonadism and congenital adrenal hypoplasia linked to a DAX-1 (NR0B1) mutation[J].Hum Reprod,2011,26(3):724-728.
[3]Loke K Y,Poh L K,Lee W W,et al.A case of X-linked adrenal hypoplasia congenita, central precocious puberty and absence of the DAX-1 gene:implications for pubertal regulation[J].Horm Res,2009,71(5):298-304.
[4]鞠长亮,王小林,乔磊,等.促性腺激素治疗男性低促性腺激素性性腺功能减退症的疗效评估[J].中国社区医师,2015,(9):14,16.
[5]Ravel C,Hyon C,Siffroi J P,et al.Are human male patients with DAX1/NR0B1 mutations infertile [J].Ann Endocrinol(Paris),2014,75(2):126-127.
Two cases of congenital adrenal hypoplasia accompanied by hypogonadotropic hypogonadism
Yuan Jinlei, Zhang Huijuan, Liu Yanxia, Liu Yanjie
(DepartmentofEndocrinology,theFirstAffiliatedHospitalofZhengzhouUniversity,Zhengzhou450052,China)
【Abstract】ObjectiveTo analyze the clinical manifestations of two cases of adrenal hypoplasia congenital(AHC) accompanied by hypogonadotropic hypogonadism(HHG), and enhance the understanding of AHC to diagnose and manage the disease more effectively.MethodsClinical features and laboratory data were collected from the patients with AHC and HHG. ResultsAHC was diagnosed by comprehensive consideration of the clinical presentations and lab tests. ConclusionPatients with AHC may also suffer from HHG, including gonadal dysgenesis and pseudo precocious puberty. AHC should be considered for patients with adrenocortical hypofunction and HHG.
【Key words】adrenal hypoplasia congenital; hypogonadotropic hypogonadism; pseudo precocious puberty
(收稿日期:2015-09-18)
【中图分类号】R 586.9
doi:10.3969/j.issn.1004-437X.2016.02.004
通讯作者:张会娟,E-mail:13598061139@163.com。
猜你喜欢
中国急救医学(2022年2期)2022-11-15
文萃报·周五版(2022年24期)2022-06-21
天津医科大学学报(2021年4期)2021-08-21
健康之家(2020年7期)2020-11-02
湖泊科学(2020年4期)2020-07-17
疯狂英语·新读写(2020年3期)2020-06-06
中国体育教练员(2017年2期)2017-07-31
河北医学(2016年5期)2016-12-01
中国民族医药杂志(2016年1期)2016-05-09
中国男科学杂志(2016年9期)2016-03-20
