
Gangliocytoma combined with a pituitary adenoma: Reports of three cases and literature review
2016-04-18 05:32ZhenminWangPengLiQiangyiZhouZhijunYangPinanLiu
Zhenmin Wang, Peng Li, Qiangyi Zhou, Zhijun Yang, Pinan Liu,2
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China
2Department of Neural Reconstruction, Beijing Neurosurgery Institute, Capital Medical University, Beijing 100050, China
Gangliocytoma combined with a pituitary adenoma: Reports of three cases and literature review
Zhenmin Wang1, Peng Li1, Qiangyi Zhou1, Zhijun Yang1, Pinan Liu1,2
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China
2Department of Neural Reconstruction, Beijing Neurosurgery Institute, Capital Medical University, Beijing 100050, China
ARTICLE INFO
Received: 20 July 2016
Revised: 18 July 2016
Accepted: 20 August 2016
© The authors 2016. This article is published with open access at www.TNCjournal.com
gangliocytoma;
Objectives:Sellar gangliocytomas are extremely rare. Since they present clinically and radiologically as pituitary adenomas, the preoperative diagnosis of these mixed tumors is very difficult. Here, we report three cases of gangliocytoma combined with pituitary adenoma and describe our findings.
1 Introduction
Gangliocytoma is a kind of slowly growing neuronal tumor with a good prognosis. Due to their mature neuronal elements, gangliocytomas were categorized under “neuronal and mixed neuronal-glial tumors”in accordance with the World Health Organization (WHO) classification of brain tumors. Their neoplastic well-differentiated neurons can be demonstrated immunohistochemically using specific neuronal markers such as neuron-specific enolase (NSE), synaptophysin, and neurofilament protein, and by electron microscopy. Because of the high degree of tumor differentiation and their low proliferative potential, gangliocytomas grow slowly and in a nonaggressive manner. Therefore, a good prognosis without recurrence can be predicted if complete tumor resection is possible.
The incidence of ganglion cell tumors lies below 4% of all brain tumors, with no sex predilection, and they are mostly located in the spinal cord, cerebral hemispheres, and brainstem. As for sellar gangliocytoma, it is extremely rare, especially co-occurring with pituitary adenoma; to date, no more than 100 cases have been reported worldwide since the first report by Greenfield in 1919[1].
Histological examination of the resected specimenhas shown areas of ganglion cells and adenomatous cells. Distinct borders were identified between gangliocytoma and adenoma cells. Most of the ganglion cells were large and mature. They contained abundant cytoplasm and Nissl granules, and were conspicuous at the periphery of the cell body. According to the electron microscopic observation, ganglion cells contained large nuclei with dense chromatin. These neurons contained numerous mitochondria, neurofilaments, and masses of ribosomes associated with the endoplasmic reticulum. Ultrastructural morphological studies also demonstrated close cell-to-cell contact between adenoma and gangliocytoma cells.
A preoperative diagnosis for this rare clinical event is very difficult since most cases present clinically and radiologically as pituitary adenomas. The definitive diagnosis of a collision sellar lesion is based on histological examination. The aim of this paper was to report our experience of collision sellar lesions in a surgical series, and to emphasize on the theories of their origin and pathogenesis along with a literature review, and then, to propose a new diagnostic clue.
2 Methods
We retrospectively studied three cases of sellar gangliocytoma associated with pituitary adenoma among 600 patients who underwent endoscopic transnasal transsphenoidal surgery between February 2003 and February 2012. This group constituted 0.5% of all pituitary adenomas in our surgical collections during the same period. Immunohistochemistry was performed for diagnosis.
3 Clinical findings
3.1 Case one
A 37-year-old woman complained of irregular menses for 3 years, and headache with a visual deficit for 1 year. Results of general physical examination and neurological examination were unremarkable. Galactorrhea was not demonstrated on pressing the breast. Neuro-ophthalmological examination disclosed bitemporal visual field defect with a slight visual acuity decline, while the fundi were normal. Magnetic resonance imaging (MRI) and computed tomography (CT) demonstrated an enlarged sella turcica containing an intrasellar mass with a suprasellar extension measuring 28 mm × 20 mm × 18 mm (Figures 1a–1f). The endocrinological evaluation revealed an elevated serum level of prolactin (PRL) of 64.83 ng/mL (reference range, 2.5–17.0 ng/mL); other parameters were within the normal range (Table 1). The patient underwent pure transnasal transsphenoidal endoscopic surgery and a tumor mass occupying the sellar cavity was completely removed. The diaphragm of the sella turcica was intact. Results of the postoperative immunohistochemical staining revealed SYN (+), CK8/18 (+), NSE (+), and GFAP (–) (Figures 1gand1h). The diagnosis by the pathologists was hypothalamic hamartoma ganglion cell tumor combined with pituitary adenoma. The postoperative period was uneventful. Endocrine investigations were performed at 3 months, 6 months, 1 year, and then annually after surgery. All hormone values were normal. At present, the patient has experienced a follow-up period of 9 years, and no signs of recurrence had been detected.
3.2 Case two
A 47-year-old man complained of acral growth, prognathism, and headache. The preoperative blood growth hormone (GH) levels were 73.6 ng/mL (normal 0–10 ng/mL,Table 1) and the diagnosis of acromegaly was made. The preoperative MRI and CT scan demonstrated an enlarged sella turcica containing an intrasellar calcified mass with a suprasellar extension measuring 31 mm × 20 mm × 16 mm (Figure 2). The mass was completely removed via the endoscopic transnasal transsphenoidal approach resulting in improvements in both clinical and laboratory results. The GH levels returned to normal. The results of postoperative immunohistochemical staining were as follows: GH (+), NSE (+), and GFAP (–) (Figure 2). The follow-up strategy was the same as the patient in case one and no abnormality has been found during the past 3 years.
3.3 Case three

Figure 1Case 1. The preoperative MRI (a–c) demonstrates an enlarged sella turcica containing an intrasellar mass with a suprasellar extension, showing high density on the CT scan (d). The postoperative MRI shows that the mass lesion was totally removed (e,f). Photomicrographs showing large, sometimes pyramid-shaped cells that are often multinucleated and undergoing atypical mitosis (g, HE, ×200), and positive expression of NSE (h, IHC, ×400). MRI: magnetic resonance imaging; CT: computed tomography; NSE: neuron specific enolase; IHC: immunohistochemistry.
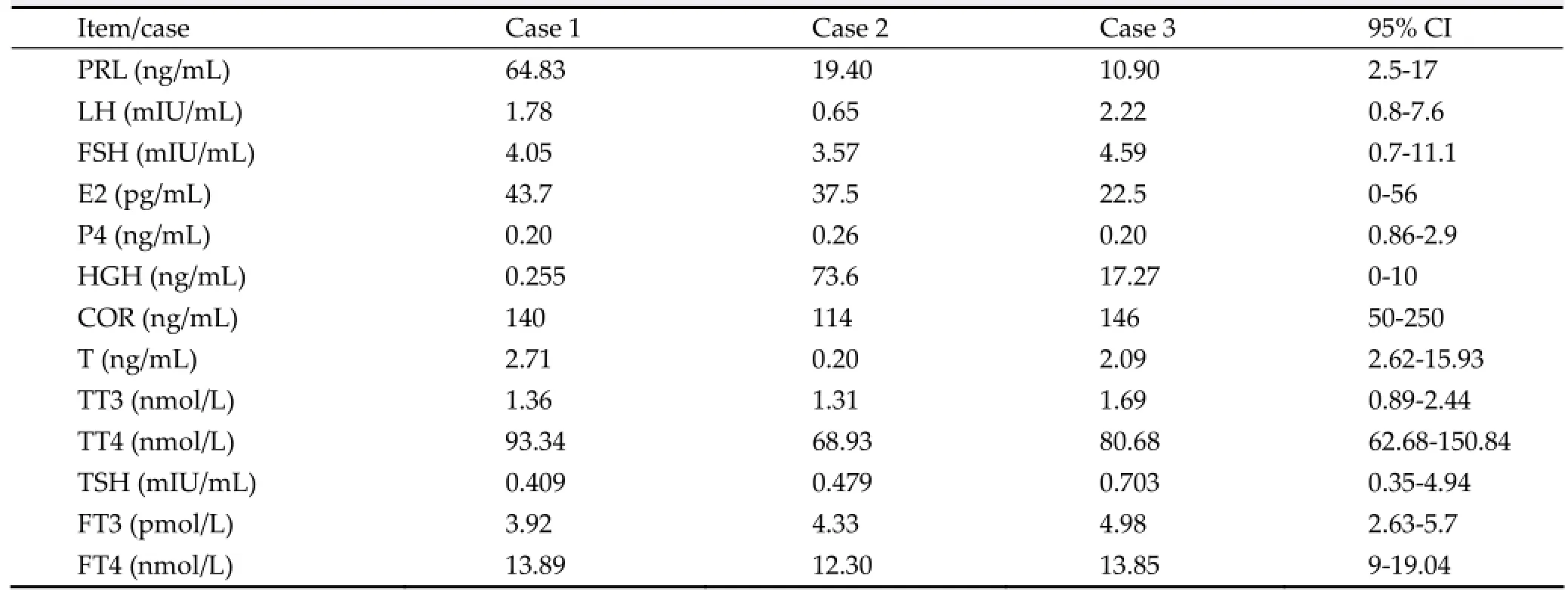
Table 1The endocrine test results of the three cases
A 46-year-old male patient complained of declined visual acuity and headache. Blood growth hormone (GH) levels were 17.27 ng/mL (normal 0–10 ng/mL,Table 1) and the diagnosis of acromegaly was made. He showed a poor response to somatostatin therapy. The preoperative MRI and CT scan demonstrated an enlarged sella turcica containing an intrasellar calcified mass with a suprasellar extension measuring 19 mm × 15 mm × 15 mm (Figure 3). Gross total removal was achieved by endoscopic transnasal transsphenoidal surgery. After surgery, both clinical and laboratory results improved; the GH and IGF-1 levels returned to normal. The results of postoperative immunohistochemical staining were as follows: GH (+), NSE (+), and GFAP (–) (Figure 3). The follow-up strategy was same as the patient in case one and no abnormality has been found during the past 30 months.
4 Discussion
Gangliocytoma associated with pituitary adenoma is a rare tumor composed of both adenomatous and gangliocytic elements. The two cell types, adenomatous and neuronal, may be admixed or may coexist in separate but adjacent tumors[2–5]. This combined tumor accounts for 0.25%–1.26% of sellar tumors[6–8]. Given the clinical and imaging similarities with pituitary adenomas, the diagnosis of a dual pathological condition of the sella is usually based on the results of histological examination.
The origin of gangliocytomas is still controversial. The theory of an incidental finding was supported by the hypothesis of abnormal migration of hypothalamic neurons within the adenohypophysial parenchyma during the early phase of embryogenesis[9]. Thus, these mixed tumors may represent an incidental concurrence of a pituitary adenoma in a pre-existing neuronal choristoma.
Another theory is that the pituitary hormone-releasing hypothalamic hormones locally produced by ganglion cells promote the adenoma formation by stimulation of adenohypophysial cells. This hypothesis is strengthened by the presence of hypophysiotropic hormones within the neurons of gangliocytomas corresponding to the relevant pituitary hormones secreted by adenoma cells[10,11]. However, lack of a correlation between the adenoma cell type and the corresponding releasing hypothalamic hormone within ganglion cells in some tumors weakens this hypothesis[8]. A third hypothesis suggests the origin of the neuronal component from the neuronal differentiation of a pre-existing pituitary adenoma, reporting the presence of transitional cell forms between neurons and adenohypophysial cells[12]. A study by Vidal et al.[13]supports the assumption of neuronal metaplasia of pituitary adenoma cells describing that somatotrophs exhibit plasticity and under certain conditions, they can undergo transdifferentiation. Moreover, the documented presence of nerve growth factor (NGF) in various adenoma cell types[14]and the evidence that NGF receptors are present in adenoma cells[15]further support this theory. Although this assumption may be challenging, we remain critical, as it is difficult to completely understand the transformation of a neoplastic pituitary cell to a well-differentiated mature neuron with the dominating embryological concepts[8].
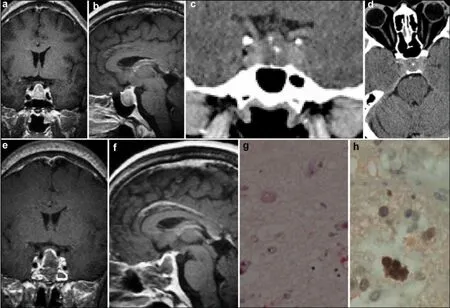
Figure 3Case 3. A sellar lesion with calcification is observed in the preoperative MRI and CT (a–d), and postoperative MRI (e–f) shows that the lesion has been removed totally. Histopathological examination showing multinucleated tumor cells (g, HE, ×200) , NSE positive expression (h, IHC, ×400). MRI: magnetic resonance imaging; CT: computed tomography; NSE: neuron specific enolase; IHC: immunohistochemistry; HE: hematoxylin and eosin.
Recently, a common origin of both adenomatous and neuronal components of the pituitary gangliocytomas has been suggested[8]. The adult pituitary gland has been proven to contain a cell population displayingcharacteristics of stem/progenitor cells[16]. Kontogeorgos et al.[8]reappraised this theory, based on the common origin of both neuronal and adenohypophysial components from uncommitted stem/progenitor cells capable of multidirectional differentiation. In this study, the authors concluded that the presence of NFP in the adenoma cell compartment of gangliocytomas indicates neuronal differentiation in adenoma cells, suggesting a common origin for neuronal and pituitary adenoma cell elements in gangliocytomas.
Previously, gangliocytomas were referred to as ganglioneuromas[17,18]. According to published studies, calcification is present in 20%–60% of ganglioneuromas[19–24]. Moreover, sellar gangliocytomas with calcification have also been detected previously[25], similar to our series (case 2 and case 3 combined with GH-secreting pituitary adenoma). As a characteristic of tumors, calcification may become a new diagnostic clue for gangliocytoma.
All three patients underwent pure endoscopic transnasal transsphenoidal surgery, and the tumors were gross totally removed. They experienced at least 30 months (30 months, 3 years, and 9 years) of the follow-up period and no signs of recurrence have been detected. Following a gross total resection, the patients received good prognosis.
Therefore, based on the theories above, we propose that calcification with GH-hypersecretion may serve as a preoperative diagnostic clue for gangliocytoma in the sella turcica.
Conflict of interests
The authors have no financial interest to disclose regarding the article.
[1] Greenfield JG. The pathological examination of forty intracranial neoplasms. Brain 1919, 42(1): 29–85.
[2] Geddes JF, Jansen GH, Robinson SF, Gömöri E, Holton JL, Monson JP, Besser GM, Révész T. ‘Gangliocytomas’ of the pituitary: A heterogeneous group of lesions with differing histogenesis. Am J Surg Pathol 2000, 24(4): 607–613.
[3] Towfighi J, Salam MM, McLendon RE, Powers S, Page RB. Ganglion cell-containing tumors of the pituitary gland. Arch Pathol Lab Med 1996, 120(4): 369–377.
[4] Morikawa M, Tamaki N, Kokunai T, Imai Y. Intrasellar pituitary gangliocyto-adenoma presenting with acromegaly: Case report. Neurosurgery 1997, 40(3): 611–614.
[5] Asada H, Otani M, Furuhata S, Inoue H, Toya S, Ogawa Y. Mixed pituitary adenoma and gangliocytoma associated with a cromegaly—Case report. Neurol Med Chir (Tokyo) 1990, 30(8): 628-632.
[6] Koutourousiou M, Kontogeorgos G, Wesseling P, Grotenhuis AJ, Seretis A. Collision sellar lesions: Experience with eight cases and review of the literature. Pituitary 2010, 13(1): 8–17.
[7] Kurosaki M, Saeger W, Lüdecke DK. Intrasellar gangliocytomas associated with acromegaly. Brain Tumor Pathol 2002, 19(2): 63–67.
[8] Kontogeorgos G, Mourouti G, Kyrodimou E, Liapi-Avgeri G, Parasi E. Ganglion cell containing pituitary adenomas: Signs of neuronal differentiation in adenoma cells. Acta Neuropathol 2006, 112(1): 21–28.
[9] Harding BCA. Malformations. In: Greenfield’s Neuropathology, 6th ed. Graham DI, Landos PL, Eds. New York: Oxford University Press, 1997.
[10] Asa SL, Scheithauer BW, Bilbao JM, Horvath E, Ryan N, Kovacs K, Randall RV, Laws ER Jr, Singer W, Linfoot JA, Thorner MO, Vale W. A case for hypothalamic acromegaly: A clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab 1984, 58(5): 796–803.
[11] Sano T, Asa SL, Kovacs K. Growth hormone-releasing hormone-producing tumors: Clinical, biochemical, and morphological manifestations. Endocr Rev 1988, 9(3): 357–373. [12] Horvath E, Kovacs K, Scheithauer BW, Lloyd RV, Smyth HS. Pituitary adenoma with neuronal choristoma (PANCH): Composite lesion or lineage infidelity? Ultrastruct Pathol 1994, 18(6): 565–574.
[13] Vidal S, Horvath E, Kovacs K, Lloyd RV, Smyth HS. Reversible transdifferentiation: Interconversion of somatotrophs and lactotrophs in pituitary hyperplasia. Mod Pathol 2001, 14(1): 20–28.
[14] Scheithauer BW, Horvath E, Kovacs K, Lloyd RV, Stefaneanu L, Buchfelder M, Fahlbusch R, von Werder K, Lyons DF. Prolactin-producing pituitary adenoma and carcinoma with neuronal components—A metaplastic lesion. Pituitary 1999, 1(3–4): 197–205.
[15] Missale C, Boroni F, Sigala S, Buriani A, Fabris M, Leon A, Dal Toso R, Spano P. Nerve growth factor in the anterior pituitary: Localization in mammotroph cells and cosecretion with prolactin by a dopamine-regulated mechanism. Proc Natl Acad Sci USA 1996, 93(9): 4240–4245.
[16] Chen JH, Hersmus N, van Duppen V, Caesens P, Denef C, Vankelecom H. The adult pituitary contains a cell population displaying stem/progenitor cell and early embryonic characteristics. Endocrinology 2005, 146(9): 3985–3998.
[17] Garrido E, Becker LF, Hoffman HJ, Hendrick EB, Humphreys R. Gangliogliomas in children. Pediatr Neurosurg 1978, 4(6): 339–346.
[18] Takahashi H, Wakabayashi K, Kawai K, Ikuta F, Tanaka R, Takeda N, Washiyama K. Neuroendocrine markers in central nervous system neuronal tumors (gangliocytoma and ganglioglioma). Acta Neuropathol 1989, 77(3): 237–243.
[19] Guo YK, Yang ZG, Li Y, Deng YP, Ma ES, Min PQ, Zhang XC. Uncommon adrenal masses: CT and MRI features with histopathologic correlation. Eur J Radiol 2007, 62(3): 359–370.
[20] Rha SE, Byun JY, Jung SE, Chun HJ, Lee HG, Lee JM. Neurogenic tumors in the abdomen: Tumor types and imaging characteristics. Radiographics 2003, 23(1): 29–43.
[21] Otal P, Mezghani S, Hassissene S, Maleux G, Colombier D, Rousseau H, Joffre F. Imaging of retroperitoneal ganglioneuroma. Eur Radiol 2001, 11(6): 940–945.
[22] Van Dyck P, Op de Beeck B, Parizel PM. Helical CT and dynamic MR features of an adrenal ganglioneuroma. JBR-BTR 2006, 89(2): 77–79.
[23] Dubois C, Jankowski A, Gay-Jeune C, Chabre O, Pasquier D, Ferretti G. Imaging of adrenal ganglioneuroma: A case report. J Radiol 2005, 86: 659–662.
[24] Lonergan GJ, Schwab CM, Suarez ES, Carlson CL. From the archives of the AFIP: Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: Radiologic-pathologic correlation1. Radiographics 2002, 22(4): 911–934.
[25] Scheithauer BW, Kovacs K, Randall RV, Horvath E, Okazaki H, Laws ER Jr. Hypothalamic neuronal hamartoma and adenohypophyseal neuronal choristoma: Their association with growth hormone adenoma of the pituitary gland. J Neuropathol Exp Neurol 1983, 42(6): 648–663.
Wang ZM, Li P, Zhou QY, Yang ZJ, Liu PN. Gangliocytoma combined with a pituitary adenoma: Reports of three cases and literature review. Transl. Neurosci. Clin. 2016, 2(3): 165–171.
* Corresponding author: Pinan Liu, E-mail: pinanliu@ccmu.edu.cn
Supported by the Youth Fund of Beijing Tiantan Hospital (Grant No.2015-YQN-10).
pituitary adenoma;
calcification;
diagnosis
Methods: The clinical data of the three cases of gangliocytoma combined with pituitary adenoma have been retrospectively analyzed, and the published literature has also been reviewed.
Results:All three patients underwent pure endonasal endoscopic surgery, and no recurrence was observed over a follow-up of at least 30 months. Growth hormone (GH)-hypersecreting adenoma and tumor calcification were detected in these mixed tumors.
Conclusions: Pure endoscopic transnasal transsphenoidal surgery may be an effective way for the treatment of this kind of tumor. Gross total resection of the tumor is recommended. In addition, calcification with GH-hypersecretion may serve as a preoperative diagnostic clue for gangliocytoma in the sella turcica.
 Translational Neuroscience and Clinics2016年3期
Translational Neuroscience and Clinics2016年3期
- Translational Neuroscience and Clinics的其它文章
- Clinical features and prognostic factors of primary intracranial malignant fibrous histiocytoma: A report of 8 cases and a literature review
- Effects of voluntary imipramine intake via food and water in paradigms of anxiety and depression in naïve mice
- Comparison of different microsurgery methods for trigeminal neuralgia
- Post-traumatic cerebrospinal fluid rhinorrhea associated with craniofacial fibrous dysplasia: Case report and literature review
- Subarachnoid hemorrhage after surgery of the medulla oblongata hemangioblastoma: A case report
- Complete resection of cavernous malformations in the hypothalamus: A case report and review of the literature