
水果和红茶中腈菌唑对映体残留的超高效合相色谱四极杆飞行时间质谱分析
2016-04-13 09:13张新忠赵悦臣罗逢健陈宗懋崔新仪楼正云
分析测试学报 2016年11期
张新忠,赵悦臣,罗逢健,陈宗懋,崔新仪,周 利,楼正云
(1.中国农业科学院茶叶研究所 农产品质量安全研究中心,浙江 杭州 310008;2.农业部茶叶产品质量安全风险评估实验室,浙江 杭州 310008;3.天津农学院 园艺园林学院,天津 300384)
水果和红茶中腈菌唑对映体残留的超高效合相色谱四极杆飞行时间质谱分析
张新忠1,2*,赵悦臣3,罗逢健1,2,陈宗懋1,2,崔新仪3,周 利1,2,楼正云1,2
(1.中国农业科学院茶叶研究所 农产品质量安全研究中心,浙江 杭州 310008;2.农业部茶叶产品质量安全风险评估实验室,浙江 杭州 310008;3.天津农学院 园艺园林学院,天津 300384)
采用超高效合相色谱四极杆飞行时间质谱,建立了手性农药腈菌唑对映体在苹果、葡萄和茶叶中的对映体拆分与残留分析方法。样品采用乙腈提取,Cleanert TPT或PestiCarb柱净化,优化合相色谱条件将腈菌唑对映体进行分离,四极杆飞行时间质谱基质外标法定量测定。对合相色谱的影响因素进行了优化,确定最佳条件为:ChromegaChrial CCA色谱柱,流动相采用CO2-异丙醇(95∶5),流速2.0 mL/min,动态背压13.79 MPa,柱温30 ℃,柱后离子化辅助溶剂为含2 mmol/L甲酸铵的甲醇-水(1∶1)溶液。结果表明:在0.01~1.00 mg/L浓度范围内,标准曲线满足线性关系,相关系数在0.98以上;在0.005,0.025,0.25 mg/kg加标水平下,苹果和葡萄中腈菌唑对映体的平均回收率(n=6)为62.5%~103.0%,相对标准偏差均不大于9.9%,方法定量下限为0.005 mg/kg;在0.01,0.05,0.5 mg/kg加标水平下,红茶中腈菌唑对映体的平均回收率(n=6)为84.1%~86.4%,相对标准偏差均小于9.6%,方法定量下限为0.01 mg/kg。
手性对映体;残留分析;腈菌唑;超高效合相色谱;四极杆飞行时间质谱
手性(Chirality)是指物质的本身立体空间结构左右对称呈镜像,但不能完全重合,是自然界本质属性之一,如同人类的左右手,相似又不能重叠,具有这种属性的异构体互为对映体(Enantiomer)。手性对映体的化学性质相似,但对映异构体在手性环境中表现出不同的生物活性、毒性毒理及降解代谢等行为,如腈菌唑(Myclobutanil,化学结构见图1),其(+)-对映体的杀菌活性显著优于(-)-对映体[1];20世纪著名的“反应停事件”也是因对映异构体性质不同所致,反应停的R-构型能很好地治疗妇女妊娠反应,而S-型则会导致婴儿四肢短小等严重的致畸作用,且不能被代谢,在反应停使用的4年期间,孕妇因服用反应停外消旋体致使1.2万名畸形“海豹婴儿”出生[2]。人体是一个由左旋氨基酸构成的手性环境,不能很好地代谢右旋分子,含右旋结构分子的物质进入人体后成为负担甚至产生损害。目前世界上常用农药中至少30%具有手性结构,且大部分以外消旋体的形式出售和使用[3],腈菌唑分子结构中含有1个手性中心,存在2个立体手性对映体,是一种高效、广谱唑类杀菌剂,其主要机制是抑制菌体麦角甾醇的脱甲基化,从而抑制菌类生长,常用于防治苹果树白粉病、葡萄炭疽病等病害,现主要以外消旋体销售。农作物在生长过程中,用于预防和防治病虫害的农药易造成在农产品中的残留,手性农药对映体施用后更容易出现对映体残留差别,从而危胁人体健康。因此手性对映体越来越引起科学家的重视[4]。目前,对虹鳟鱼[5]、黄瓜[6]、蚯蚓[7]、水[8- 9]和土壤[6-9]中腈菌唑手性对映体的残留分析已有报道,采用的方法有气相色谱质谱法[5]、液相色谱法[9]及液相色谱-串联质谱法[6-8],但传统的气相色谱、液相色谱法对载气、流动相的要求高,溶剂使用量大,对环境污染影响大,且方法开发的复杂性高,而合相色谱能更好地解决这些问题。
近些年基于超临界流体色谱原理发展起来的合相色谱技术(Convergence chromatography),以超临界流体状态CO2为主要流动相,辅以一定比例的有机溶剂(醇或酸等)作为改性剂,以实现化合物的分离分析[10]。合相色谱综合了超临界流体色谱(SFC)和液相色谱(LC)的优点,介于气相色谱和液相色谱技术之间,分析条件温和、不受样品挥发性的限制,其固定相可采用现有的正相、反相固定相材料,既可分析不适宜气相色谱分析的高沸点、低挥发性样品,又能对普通液相色谱难分离的结构类似物[11-14]、手性化合物[15- 17]等进行分离分析。四极杆-飞行时间质谱(Q-TOF/MS)能提供高分辨率精确质荷比离子用于化合物的定性定量分析,同时利用串联质谱的功能进行二级质谱裂解可获得大量高分辨的质谱碎片离子信息,亦可用于进一步结构确定和定量分析[18]。本课题组最早应用飞行时间质谱对手性对映体进行残留分析,前期采用手性色谱柱结合高效液相色谱四极杆飞行时间质谱[19]、合相色谱四极杆飞行时间质谱[16]分别建立了茶叶等基质中氟环唑等手性农药对映体的残留分析方法,为此后的研究奠定了基础。
为扩展合相色谱的应用范围,本文采用乙腈提取,固相萃取柱净化苹果、葡萄和茶叶,超高效合相色谱(UPC2)结合手性色谱柱对腈菌唑对映体进行拆分,四极杆-飞行时间质谱(Q-TOF/MS)技术提取精确质荷比离子进行定量,二级质谱碎片辅助定性,建立了苹果、葡萄和茶叶中腈菌唑残留的分析方法,方法准确可靠,能够满足残留分析的要求[20]。
1 实验部分
1.1 仪器与试剂

乙腈、甲醇、异丙醇(色谱纯,美国Honeywell公司);甲酸、甲酸铵、乙酸铵(色谱纯,德国CNW科技有限公司);无水硫酸钠、氯化钠(分析纯,上海市四赫维化工有限公司);纯净水(杭州娃哈哈有限公司)。
腈菌唑标准品(98%,德国Dr.Ehrenstorfer GmbH)。苹果、葡萄、红茶空白样品购于市场,经检测不含腈菌唑残留。
1.2 实验方法
将新鲜的苹果、葡萄果肉果皮分离后,分别用粉碎机粉碎(茶叶直接用粉碎机粉碎)。称取磨碎后的样品10.0 g(茶叶称取5.00 g)于50 mL离心管中,加入5 mL纯净水,旋涡1 min使样品充分润湿,静置30 min,再加入20 mL乙腈,均质2 min后静置过夜,加入5 g NaCl振荡涡旋混匀后,超声提取10 min,5 000 r/min离心5 min,取乙腈层10 mL至圆底烧瓶,45 ℃真空旋转浓缩至近干,加入5 mL乙腈超声溶解。在500 mg/6 mL Cleanert PestiCarb固相萃取柱(茶叶用2 000 mg/12 mL Cleanert TPT 固相萃取柱)上加入约2 cm高的无水硫酸钠,依次用5 mL乙腈-苯(3∶1)和5 mL乙腈预淋洗,待液面降至Na2SO4顶部时,将样品提取液转移至柱上,弃去流出液,再用5 mL乙腈洗涤圆底烧瓶并转移至柱上,接收流出液至鸡心瓶,随后加入乙腈-苯(3∶1)洗脱,共接收20 mL,在45 ℃水浴中旋转浓缩近干,N2吹干后,加入1.0 mL乙腈涡旋超声定容溶解,过0.22 μm滤膜至进样瓶,UPC2-QTOF/MS进样5 μL,基质外标法定量。
1.3 色谱-质谱条件
色谱柱:ChromegaChrial CCA柱(15 cm×4.6 mm,5 μm,美国ES Industries公司);柱温30 ℃:进样量5 μL;样品盘温度10 ℃;流动相:CO2-异丙醇(95∶5)等度洗脱30 min;背压13.79 MPa;流速2.0 mL/min。
质谱条件:ESI+-QTOF/MS全扫描模式,提取腈菌唑准分子离子峰[M+H]+m/z(289.121 5±10)ppm定量;毛细管喷雾电压3 kV;锥孔电压40 V;源补偿电压80 V;源温度120 ℃;脱溶剂化温度350 ℃;锥孔气N2,流速50 L/h;雾化气N2,流速600 L/h;离子化辅助溶剂:含2 mmol/L甲酸铵的甲醇-水(1∶1),流量0.20 mL/min;质量扫描范围m/z150~600;采集速率2 spectra/s;实施校准参比离子m/z556.277 1。
1.4 标准工作溶液的配制与标准曲线
称取0.010 g腈菌唑标准品至50 mL容量瓶中,用乙腈稀释定容得200 mg/L储备液(对应其对映单体的浓度为100 mg/L),-20 ℃冰箱存放。用乙腈稀释配成对映单体浓度依次为1.0,0.50,0.25,0.10,0.05,0.025,0.010 mg/L的系列溶剂,用经过提取的空白基质溶液配制相应浓度的5种基质标准溶液,采用UPC2-QTOF/MS进样5 μL,每个样品进样3次,以腈菌唑对映体浓度为横坐标(x,mg/L),3次进样提取离子峰面积的平均值为纵坐标(y)进行曲线拟合,获得腈菌唑对映体的溶剂和基质标准曲线方程、相关系数与基质效应。
1.5 回收率与相对标准偏差
在不含腈菌唑的空白苹果肉、苹果皮、葡萄肉、葡萄皮和红茶样品中,分别加入2.50,0.50,0.05 mg/L的标准溶液1 mL,旋涡均匀,分别相当于0.25,0.05,0.005 mg/kg 3个添加浓度,静置2 h。按照“1.2”方法处理,每个水平重复6次。同时按“1.4”方法配制相应浓度的基质标准溶液,基质外标法定量,获得加标回收率、相对标准偏差。
2 结果与讨论
2.1 色谱条件优化
在ChromegaChrial CCA柱下,针对不同流动相(乙腈、甲醇、异丙醇)改性剂、不同柱温(15~50 ℃)、不同流速(1.2~3.2 mL/min)以及不同背压(31.03~10.34 MPa)下,进行色谱条件优化。
2.1.1 流动相改性剂比例的优化 UPC2分析时,使用非极性CO2超临界流体为流动相,加入改性剂可以改变其对手性化合物不同对映体之间的洗脱能力,使手性化合物对映体的峰形、保留时间及拆分效果得到改善。分别以乙腈、甲醇和异丙醇作为改性剂,在流速2.0 mL/min,背压17.24 MPa和柱温25 ℃条件下,初步比较不同比例(20%,15%,10%,5%,3%,0%)的改性剂对腈菌唑手性对映体分离效果的影响。结果表明:使用乙腈和甲醇作为改性剂时,均无法拆分腈菌唑对映体;以5%异丙醇为改性剂时,峰1和峰2所对应腈菌唑的保留时间分别为16.17 min和24.85 min,分离度为6.22,达到完全分离,而当异丙醇比例为3%时,在30 min分析时间内不能获得完整的对映体峰,故初步确定以5%异丙醇作为改性剂。
2.1.2 流速对分离的影响 UPC2分析时,使用CO2作为流动相,其在超临界状态下具有低粘度和高扩散系数的特性,但流动相流速会影响对映体的分离度、出峰时间及色谱柱压力。本文在4%异丙醇,柱温25 ℃,背压17.24 MPa的条件下,考察了流速在1.2~3.2 mL/min范围内对1 mg/L标准溶液分离的影响,结果见表1。结果表明:流速降低,保留时间延长,峰形展宽程度高,在一定范围内对映体分离度增大、柱压降低,为使对映体得到很好的分离,同时保留时间和色谱柱压合适,选择流速为2.0 mL/min。

表 1 不同流速下腈菌唑对映体的保留时间t、容量因子k、分离因子α和分离度RTable 1 Retention times(t),capacity factors(k),separation factors(α) and resolutions(R) of myclobutanil enantiomers at different flow rates
2.1.3 动态背压的优化 CO2处于超临界流体状态下时,不同压力下其对化合物的溶解能力不同,溶解度随CO2的增多而增大。在整个运行过程中保持超临界流体状态CO2的动态背压,是影响分离过程的重要因素之一。因此在4%异丙醇,柱温25 ℃,流速2 mL/min的条件下,进样1 mg/L标准溶液,考察动态背压(10.34~31.03 MPa)对腈菌唑对映体分离的影响。结果表明:动态背压升高会导致系统柱压背压升高,腈菌唑对映体的保留时间减少,t1和t2分别由32.68,52.26 min缩短至14.35,22.62 min;容量因子降低,k1和k2分别由32.69,52.87降至13.79,22.33;分离时间缩短,分离因子α总体变化不大;对映体分离度R降低,由6.50降至5.19,见图2A。综合考虑,最终选择动态背压为13.79 MPa。
2.1.4 色谱柱温的选择 根据热动力学原理,色谱柱温度对化合物的保留时间和分离影响很大。在4%IPA,背压13.79 MPa,流速2 mL/min的条件下,对1 mg/L溶剂标准溶液进样,考察了柱温(15~50 ℃)对拆分效果的影响(图2B)。结果表明:随着柱温升高,对映体保留时间延长,t1和t2分别由23.39,36.94 min延长到31.70,53.43 min;容量因子增加,k1和k2分别由23.11,37.08增加到31.68,54.08;分离因子变化较小,α总体呈上升趋势,由1.60增至1.71;分离度R在40 ℃表现出最大值(R=7.27)。但由于合相色谱以超临界CO2为主要流动相,随着色谱柱温度的增高,超临界流体CO2密度减小,其溶剂化能力降低,从而使化合物的保留时间增长,为了降低腈菌唑的保留时间,同时考虑各方面的影响,故本实验选择30 ℃作为柱温。
2.1.5 辅助溶剂的选择 合相色谱中,由于大量流动相为CO2,CO2被引入质谱离子源时,离子化响应较差。因此需在柱后加入辅助溶剂来促进待测化合物的离子化程度,提高质谱响应。对比研究了0.20 mL/min流量下,不同辅助溶剂,即含0.1%~1%甲酸的乙腈、含0.1%~2%甲酸的甲醇、含2~10 mmol/L甲酸铵的甲醇、含2~10 mmol/L乙酸铵的甲醇、含2~10 mmol/L甲酸铵的甲醇-水(1∶1)以及含2~10 mmol/L乙酸铵的甲醇-水(1∶1)对1 mg/L腈菌唑对映体质谱响应的影响。结果表明:当以含2 mmol/L甲酸铵的甲醇-水(1∶1)作为辅助溶剂时,腈菌唑[M+H]+离子响应高于其它辅助溶剂,这可能是因为,腈菌唑在质谱中主要产生[M+H]+离子,在甲醇、水都存在的条件下,易发生电解(CH3OH+H2O→CO2+6H+)[21],产生更多的H+[22-23],从而提高了腈菌唑的离子化效率,并通过竞争效应抑制了[M+Na]+的产生[24]。因此,最终选择含2 mmol/L甲酸铵的甲醇-水(1∶1)作为柱后离子化辅助溶剂。
综合上述优化条件,最终选择CCA柱下,流动相为CO2-异丙醇(95∶5),流速2.0 mL/min,动态背压13.79 MPa,柱温30 ℃,辅助溶剂为含2 mmol/L甲酸铵的甲醇-水(1∶1)作为仪器分析条件。在该条件下,腈菌唑对映体的分离度R>1.5,完全满足分析要求。
2.2 提取与净化方法的优化
考虑到基质的含水量、组分复杂程度、受药和食用部位的差别,选择苹果、葡萄和茶叶作为研究基质。首先苹果、葡萄的含水量大,而茶叶的含水量小,农药残留提取难度有差别;其次苹果、葡萄的含糖量较高,而茶叶中含咖啡碱、茶氨酸等组分,基质复杂程度高,导致净化难度不同;再次,腈菌唑多采用叶面喷雾方法施药,茶叶冲泡饮用加工后叶片,而苹果、葡萄在生长过程中多是套袋处理,且苹果为木本植物、葡萄为藤本植物,其果型大小和种植管理方式有差异,导致叶片、果皮、果肉中农药残留有差异;同时人们食用苹果、葡萄时,一般会去皮处理,故本文将苹果、葡萄的皮和肉分离,分别进行分析。而农药残留分析中,基质越复杂,方法建立难度越大,因此本文选择红茶为基质代表,优化提取净化方法。在中等添加浓度下,对比QuEChERS方法和《茶叶中农药及相关化学品残留量的测定液相色谱-串联质谱 GB23205-2008》,发现QuEChERS方法的回收率略高于国标方法,但QuEChERS方法的浓缩系数不够,低浓度无法检测,因此最终选择在国标方法的基础上,对样品前处理方法进行改进,以保证获得更好的回收率。
2.2.1 提取方法的优化 对茶叶中残留的农药进行提取时,加水润湿茶叶与不加水对茶叶中农药的提取效果有差别,通常对于弱极性农药提取率的影响很小,而对于强极性的农药影响显著[19,25]。本文比较了不加水与加水条件下,乙腈对茶叶中腈菌唑提取率的影响,分别对乙腈、乙腈-水提取后浓缩的样品,对比了向圆底烧瓶中加入5 mL不同溶液(水、乙腈-水(1∶1)、乙腈、乙腈-苯(3∶1))来超声溶解残余物上样净化的结果。结果表明:乙腈提取的样品,4种溶液溶解上样的回收率分别为88.6%,109.4%,112.9%和93.4%;乙腈-水提取的样品,4种溶液溶解上样的回收率分别为70.5%,84.7%,106.0%和106.5%。由于加水提取时,茶叶中的农药残留更容易提取出来,但杂质也被提取,浓缩后附着在瓶壁上,当溶解上样液含有水时(水、乙腈-水),杂质溶解下来,增加了后续净化难度,导致回收率降低;当上样液不含水时(乙腈、乙腈-苯),可保证较好的回收率。故对茶叶样品,采用乙腈-水提取,浓缩后,乙腈溶解上样,既能提高回收率,且能保证后续净化效果。
2.2.2 净化方法优化 在TPT柱上,利用标准溶液上样时,分别采用乙腈、乙腈-苯(3∶1)进行洗脱20 mL,分段接收,每段2 mL,测定各段回收率。结果表明:以乙腈洗脱时,腈菌唑在0~4,4~6,6~8,8~10,10~12,12~14,14~16 mL的回收率分别为10.3%,7.8%,30.0%,35.5%,22.9%,9.1%和10%;而以乙腈-苯(3∶1)洗脱时,腈菌唑在0~4,4~6,6~8,8~10,10~12,12~14,14~16 mL的回收率分别为3.9%,22.0%,44.9%,24.3%,7.3%,6.6%和2.3%,此时腈菌唑主要在4~14 mL被洗脱下来,回收率总计达105.1%,表明腈菌唑的洗脱阶段更为集中,洗脱液体积的减小能减少杂质的共流出。因此实验选择乙腈5 mL作为上样液和淋洗液,乙腈-苯(3∶1)洗脱16 mL接收,在减少杂质的共流出时,确保高回收率。而苹果和葡萄基质较茶叶简单,采用500 mg/6 mL Cleanert PestiCarb固相萃取柱即可满足净化的需求。
2.3 标准曲线、相关系数与基质效应
在优化条件下,按“1.4”方法考察腈菌唑对映体的溶剂和基质标准曲线方程、相关系数,结果见表2。结果表明,在0.010 ~1.0 mg/L浓度范围内线性关系良好,相关系数(r)均在0.98以上,在最低进样浓度下,根据S/N=3计算,方法的检出限(LOD)为0.10~0.46 μg/kg(见表2)。
以腈菌唑对映体在基质中的线性方程斜率与其在乙腈中的线性方程斜率的比值考察基质效应,结果表明:在苹果皮、葡萄肉中表现出一定的基质增强效应,而在苹果肉中的基质效应不明显,在葡萄皮和红茶中表现出较强的基质减弱效应。因此本方法采用基质标准溶液进行定量分析。

表2 腈菌唑对映体在不同样品基质中的线性范围、线性方程、相关系数(r)和基质效应结果Table 2 Linear ranges,regression equations,correlation coefficients(r) and matrix effects for myclobutanil enantiomers in different matrices
2.4 加标回收率、相对标准偏差与定量下限
按照“1.2”方法处理进行3个浓度水平的样品加标回收实验,每个水平重复6次。同时按“1.3”方法配制相应浓度的基质标准溶液,基质外标法定量,回收率和相对标准偏差见表3。结果表明:在苹果肉中的平均回收率为80.3%~93.6%,相对标准偏差(RSD)为3.3%~8.8%;在苹果皮中的平均回收率为62.5%~103.0%,RSD为2.0%~7.8%;在葡萄肉中的平均回收率为75.8%~87.2%,RSD为2.9%~7.7%;在葡萄皮中的平均回收率为75.8%~93.1%,RSD为6.6%~9.9%;在红茶中的平均回收率为84.1%~86.4%,RSD为3.5%~9.6%。以最低添加浓度计算方法的定量下限,得苹果、葡萄基质中腈菌唑对映体的定量下限为0.005 mg/kg;红茶基质中腈菌唑对映体的定量下限为0.01 mg/kg。图3为葡萄皮的空白样品、基质标准样品和添加浓度样品的提取离子特征色谱图。
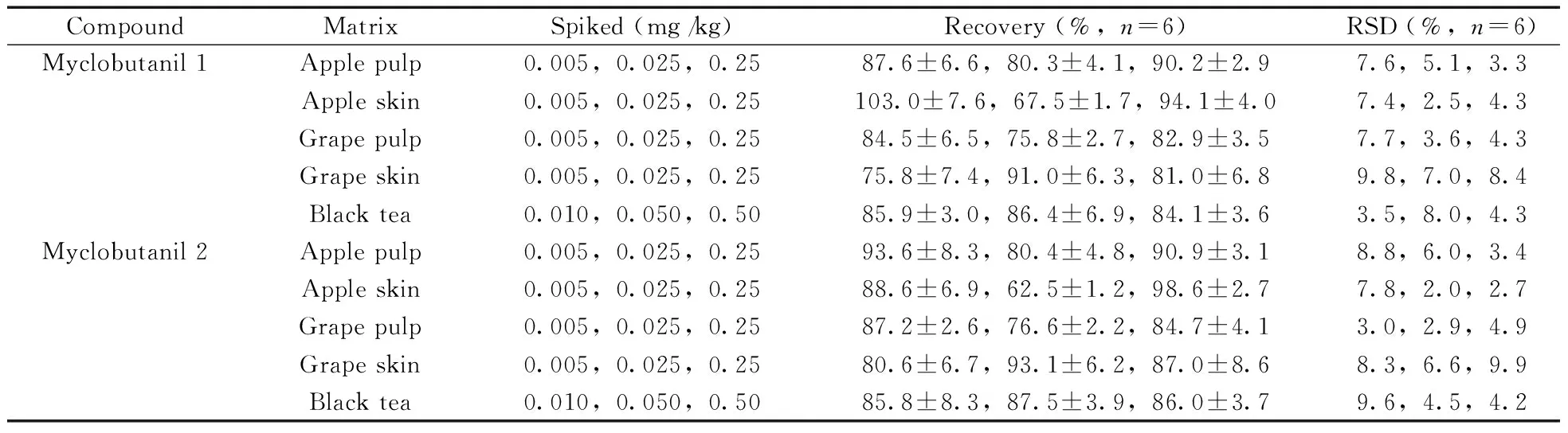
表3 腈菌唑对映体在不同基质中的加标回收率与相对标准偏差Table 3 Average spiked recoveries and relative standard deviations(RSDs) for myclobutanil enantiomers in different matrices

图3 葡萄皮的空白样品(A)、基质标准样品(B)和对应添加浓度样品(C)的提取离子色谱图
Fig.3 Chromatograms of the blank sample(A),matrix standard sample(B),and the spiked sample(C) of myclobutanil enantiomers in grape skin
2.5 方法的应用
采用本方法检测市售苹果、葡萄和红茶样品各20份,均未检出腈菌唑对映体残留。
3 结 论
本文利用合相色谱结合ChromegaChrial CCA色谱柱实现了腈菌唑对映体的分离。样品采用乙腈-水混合溶液提取,乙腈上样淋洗,乙腈-苯(3∶1)过Cleanert TPT或PestiCarb柱净化洗脱,超高效合相色谱-四极杆飞行时间质谱(UPC2-QTOF/MS)基质外标法分析,建立了手性农药腈菌唑对映体在苹果、葡萄和红茶中的对映体拆分与残留检测方法。本方法结果稳定可靠,能够满足残留分析的需要,可用于实际样品的检测。
[1] Deng Z B,Hu J Y.Pesticides(邓朱波,胡继业.农药),2011,50(5):371-373.
[2] Wang D,Fan Q H.Sci.Technol.Daily(王东,范青华.科技日报),2001-10-17,9.
[3] Ulrich E M,Morrison C N,Goldsmith M R,Foreman W T.Rev.Environ.Contam.Toxicol.,2012,217:1-74.
[4] Liu W P,Gan J Y,Schlenk D,Jury W A.Proc.Natl.Acad.Sci.USA,2005,102:701-706.
[5] Konwick B J,Garrison A W,Avants J K,Fisk A T.Aquat.Toxicol.,2006,80:372-381.
[6] Dong F S,Cheng L,Liu X G,Xu J,Li J,Li Y B,Kong Z Q,Jian Q,Zheng Y Q.J.Agric.FoodChem.,2012,60:1929-1936.
[7] Chen J H,Wang H L,Guo B Y,Li J Z.Int.J.Environ.Anal.Chem.,2014,94(8):791-800.
[8] Li Y B,Dong F S,Liu X G,Xu J,Han Y T,Zheng Y Q.Chemosphere,2015,122:145-153.
[9] Tian Q,Zhou Z Q,Lv C G,Huang Y,Ren L P.Anal.Methods,2010,2:617-622.
[10] Rao G V N,Gnanadev G,Ravi B,Dhananjaya D,Manoj P,Indu B,Nadh R V.AnalMethods,2013,5:4832-4837.
[11] Zhou W,Wang B,Liu Q Q,Yang S X,Wang L T.Chin.J.Anal.Chem.(周围,王波,刘倩倩,杨盛鑫,王丽婷.分析化学),2015,43(1):115-120.
[12] Zhou Q,Gao B Y,Zhang X,Xu Y W,Shi H M,Yu L L.FoodChem.,2014,143:199-204.
[13] Li B,Zhao H Y,Liu W,Fan S,Li L P,Wu G H,Xue Y,Zhao R.J.Instrum.Anal.(李兵,赵海燕,刘伟,范赛,李丽萍,吴国华,薛颖,赵榕.分析测试学报),2015,34(7):813-818.
[14] Lin C H,Yan N,Xu Z H,Liao W L,Fan N L,Yang S M.J.Instrum.Anal.(林春花,严楠,许招会,廖维林,范乃立,杨绍明.分析测试学报),2014,33(11):1322-1326.
[15] Pan X L,Dong F S,Xu J,Liu X G,Chen Z L,Zheng Y Q.J.Hazard.Mater.,2016,311:115-124.
[16] Zhao Y C,Zhang X Z,Luo F J,Zhou L,Chen Z M,Cui X Y.Chin.J.Anal.Chem.(赵悦臣,张新忠,罗逢健,周利,陈宗懋,崔新仪.分析化学),2016,44(8):1200-1208.
[17] Wang B,Yan H,Wang S J,Liu A J,Yang S X,Zhou W.J.Instrum.Anal.(王波,闫衡,王肃军,刘阿静,杨盛鑫,周围.分析测试学报),2015,34(7):824-828.
[18] Park J S,Jung M Y.J.Agric.FoodChem.,2012,60:10015-10026.
[19] Zhang X Z,Luo F J,Lou Z Y,Lue M L,Chen Z M.J.Chromatogr.A,2014,1359:212-223.
[20] General Administration of Quality Supervision,Inspection and Quarantine of the People's Republic of China.ResiduesAnalysisQualityControlGuide.Beijing,(中国人民共和国质量监督检验检疫总局.残留分析质量控制指南.北京),2002.
[21] Cloutier C R,Wilkinson D P.Int.J.HydrogenEnergy,2010,35:3967-3984.
[22] Peng Y,Zhang S,Gong X,Ma X,Yang C,Zhang X.Anal.Chem.,2011,83:8863-8866.
[23] Fisher C M,Kharlamova A,McLuckey S A.Anal.Chem.,2014,86:4581-4588.
[24] Verkerk U H,Kebarle P.J.Am.Soc.MassSpectrom.,2005,16:1325-1341.
[25] Chen H P,Liu X,Wang C P,Wang Q H,Jiang Y.Chin.J.Anal.Lab.(陈红平,刘新,王川丕,汪庆华,蒋迎.分析试验室),2011,30(8):48-53.
Determination of Myclobutanil Enantiomers Residues in Fruit and Black Tea Samples by Ultra Performance Convergence Chromatography Quadrupole Time-of-Flight Mass Spectrometry
ZHANG Xin-zhong1,2*,ZHAO Yue-chen3,LUO Feng-jian1,2,CHEN Zong-mao1,2,CUI Xin-yi3,ZHOU Li1,2,LOU Zheng-yun1,2
(1.Research Center of Quality Safety for Agricultural Products,Tea Research Institute,Chinese Academy of Agricultural Sciences,Hangzhou 310008,China;2.Tea Product Quality and Safety Risk Assessment Laboratory,Ministry of Agriculture,Hangzhou 310008,China;3.College of Horticulture and Landscape,Tianjin Agricultural University,Tianjin 300384,China)
A separation and residue determination method for myclobutanil enantiomers in apple,grape and black tea was firstly developed by ultra performance convergence chromatography combined with quadrupole time-of-flight mass spectrometry (UPC2-QTOF/MS).Samples were extracted with acetonitrile,purified with Cleaneert TPT or Cleanert PestiCarb solid-phase extraction(SPE) columns,and separated in the optimum condition of convergence chromatography,then analyzed by UPC2-QTOF/MS with the matrix external standard method.All the influence factors of convergence chromatography(chromatographic column,mobile phase modifier and proportion,column temperature,automated backpressure regulator,and post-column auxiliary solvent) were optimized.The best conditions were as follows:ChromegaChrial CCA column with a mobile phase of CO2-isopropanol(95∶5),a flow rate of 2.0 mL/min,an automated backpressure regulator(ABPR) of 13.79 MPa,a column temperature of 30 ℃,and a post-column auxiliary solvent of methanol-water(1∶1) containing 2 mmol/L ammonium formate.The results showed that the linear ranges of myclobutanil enantiomers were in the range of 0.01-1.00 mg/L,and the correlation coefficients were above 0.98.The recoveries of myclobutanil enantiomers at three spiked levels of 0.005,0.025,0.25 mg/kg in fruit matrix were in the range of 62.5% - 103.0% with relative standard deviations (RSDs,n=6) not more than 9.9%,and the limits of quantitation(LOQ) of enantiomers were 0.005 mg/kg.The recoveries of myclobutanil enantiomers at three spiked levels of 0.01,0.05,0.5 mg/kg in black tea matrix were 84.1% - 86.4% with RSDs(n=6) less than 9.6%,and the LOQ for these two enantiomers were 0.01 mg/kg.The method was rapid,convenient and reliable,and could meet the requirement for residue analysis.
chiral enantiomers;residual analysis;myclobutanil;ultra performance convergence chromatography(UPC2);quadrupole time-of-flight mass spectrometry(QTOF/MS)
2016-05-09;
2016-06-19
国家自然科学青年基金(21107137);国家茶叶产业技术体系(CARS-23);浙江省自然科学基金(Y3100259);中国农业科学院创新工程
10.3969/j.issn.1004-4957.2016.11.002
O657.63;S482.2
A
1004-4957(2016)11-1376-08
*通讯作者:张新忠,博士,副研究员,研究方向:农药残留色谱质谱分析与风险评估,Tel:0571-87963516,E-mail:zxz.1982@163.com
猜你喜欢
煤化工(2022年3期)2022-07-08
现代仪器与医疗(2022年1期)2022-04-19
日用电器(2022年3期)2022-04-14
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
中国土壤与肥料(2021年5期)2021-12-02
现代仪器与医疗(2021年2期)2021-07-21
今日农业(2020年22期)2020-12-14
分析化学(2018年12期)2018-01-22
食品界(2017年7期)2017-08-24
山东工业技术(2016年10期)2016-09-06
