
一种高效简便的原代神经元培养方法
2016-04-11 05:46:35张涛胡怀强王旭辉牛兵陶珍曹秉振
神经损伤与功能重建 2016年6期
张涛,胡怀强,王旭辉,牛兵,陶珍,曹秉振
·论著·
一种高效简便的原代神经元培养方法
张涛1、2,胡怀强1,王旭辉3,牛兵1,陶珍1,曹秉振1
目的:建立一种简便高效的原代神经元培养方法。方法:出生12 h内的Wistar乳鼠,分离皮质后剪碎,0.25%胰酶消化30 min,漂洗后轻柔吹散细胞,过滤后1 000 rpm离心5 min,弃上清,加接种液吹匀后过滤,静置3~5 min,取上层液体计数并接种。接种后4、45、96 h全量换液,之后每3 d半量换液1次。培养第5天评估神经元,NSE染色、NMDAR1染色和台盼兰染色。结果:神经元纯度高(>95%),死亡率低(<5%),细胞形态好,交联充分,背景干净。结论:利用新生大鼠皮质建立了高质量、高产量、简便的神经元培养模型。
皮质神经元;原代培养;细胞模型;乳鼠
神经元原代培养是研究神经系统疾病的重要基础模型[1,2]。目前的模型大多存在一些瑕疵,如手术复杂,手术时间长,影响神经元活力;神经元纯度高,但死亡率也较高;价格昂贵,对实验器材及相关试剂要求高[3-5]等。本课题组采用新生乳鼠皮质,建立了一种简单、廉价、高产、高纯度、高成活率、低背景的神经元培养模型。
1 材料与方法
1.1 材料
1.1.1 实验动物与试剂 新生12 h内Wsitar乳鼠,雌雄不拘,购自山东大学实验动物中心。0.25%胰酶(含EDTA)(PYG0015)购于武 汉 博 士 德 公 司 ,Neurobasal-A(108880-022)、B27(17504-044)、glutamax(35050-61)、Hanks’Balanced Salt Solution(HBSS)液(14175-079)、小 牛 血 清(16000-044)购于Gibco公司,DMEM/F12(SH30023)购 于 Hyclone公 司 ,DnaseI(D8071)购于上海联硕公司,抗体NSE(neuron-specific enolase)(SAB4200571)、多聚赖氨酸(P1399)购于Sigma公司,抗体NMDAR1(N methyl D aspartate recptor(ab134308)购于abcam公司。
1.2 方法
1.2.1 解剖及培养 少量水合氯醛捂鼻麻醉乳鼠,浸入75%酒精,再浸入无菌冰水中约2 min。无菌冰台上迅速完整分离整个脑组织,置于冰HBSS液中漂洗。首先剪去尾侧小脑,再沿矢状线剪开两侧大脑,将脑组织移入另一干净的冰HBSS液中。小眼科镊去除掉皮质下脑干、间脑等组织,完整剥离软脑膜,去除残留血管、嗅球等组织。取出皮质放入接种液PM(plantation medium,10% FBS/DMEM/F12)中,剪碎。将剪碎的脑组织吸入37℃预热的胰酶中37℃消化30 min。将絮状的皮质组织吸入PM液中放置3 min×2次。再将皮质组织吸入吹打液BM(blowing medium,0.2 mg/mL DnaseI/10%FBS/DMEM/F12)中,用塑料一次性无菌吸管轻柔缓慢吹打,吹打前将管口稍微过火钝化,吹打时避免吹入气泡,吹打BM液为米汤状。200目细胞筛过滤后1 000 rpm离心5 min,弃上清后加入适量PM液稍微吹匀后再次200目细胞筛过滤,静置约3 min后取上清液计数细胞密度,计算好接种浓度后添加适量PM液配好接种液。接种量:9.5×104/孔(24孔板),7.0×105/孔(6孔板)。接种后4、45、96 h使用NM(neurobasal meduim,含Neurobasal-A,B27,glutamax,青链霉素)全量换液,每次换液前可用HBSS液轻柔漂洗1次,之后可每3天半量换液1次。
1.2.2 细胞鉴定 培养第5天评估神经元,光镜下观察神经元并行NSE、NMDAR1免疫细胞化学染色和台盼兰染色。免疫细胞化学染色:4%多聚甲醛室温下固定30 min,PBS液漂洗,3%H2O2去离子水10 min,PBS液漂洗后山羊血清封闭液20 min,NSE或NMDAR1孵育4℃过夜,漂洗后生物素标记山羊抗大鼠IgG室温15 min,漂洗后辣根酶标记链霉卵白素室温15 min,漂洗后DAB显色。台盼兰染色:400 μL的2%台盼兰染液/孔(24孔板),室温下12 min,倒掉染液,倒置光学显微镜观察计数。
2 结果
2.1 神经元生长情况变化
接种约12 min左右,大部分神经元即沉底,但贴壁不牢。接种4 h可见少数神经元长出突起,神经元体积较小。接种第1天,大部分神经元长出突起。接种第2天,几乎所有神经元均长出突起,突起较长。部分非神经元细胞、死亡神经元、组织碎屑可在45 h全量换液时去除。换液后神经元纯度高,活力好,背景干净。接种第3天开始,神经元形态基本成熟,体积较前变大。接种第4天神经形态进一步发育成熟,细胞饱满透亮,突起交联充分。部分活力较差的神经元和死亡细胞在接种96 h全量换液时可去除。此后死亡细胞仍贴壁牢,再全量换液无类似明显效果。第5~9天细胞状态无明显变化,可用于相关实验。第10天神经元逐渐衰老,透亮度下降,部分神经元固缩凋亡,见图1。
2.2 神经元状态评估鉴定
接种第5天,神经元特异性标志物NSE染色阳性率>95%,见图2A;NMDAR染色可见神经元突起细长,交联密布充分,见图2B;台盼兰染色阳性率<5%,见图1C、D。神经元生长状态好,达到进一步实验要求。
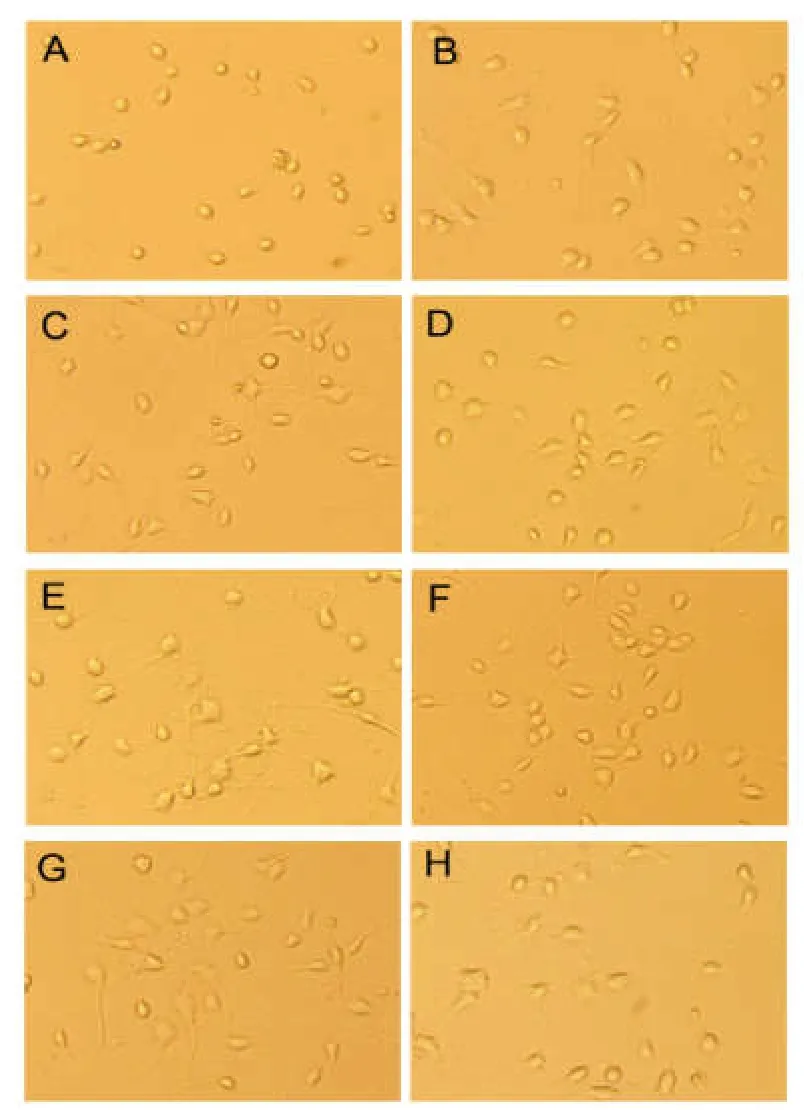
图1 培养的皮质神经元生长情况(光镜,×200)
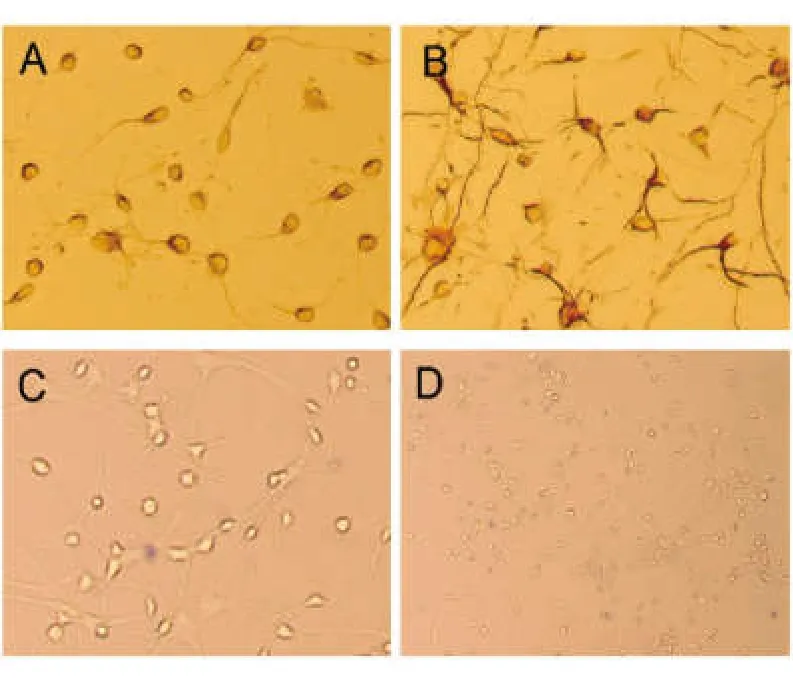
图2 神经元评估鉴定
3 讨论
神经元的培养模型很多,还有一些实验[6,7]通过诱导其它细胞分化为神经元。
细节的处理是决定神经元培养效果的关键。本研究主要进行了以下调整:①早期全量换液。本课题组研究发现全量换液对细胞活力的影响不明显,而且可以清除活力差的细胞、非神经元细胞和坏死碎屑。要注意操作温柔、快速、一孔一换液,减少干底时间和污染。②换液时间很重要。采用NM液于接种后第4、45、96 h全量换液。能清洁神经元,保持神经元高活力、高纯度、低背景。换液后超过2 d或接种第4天以后,神经元已经贴壁非常牢固,此时全量换液并不能将死亡的神经元全部换掉。常规接种后每3天半量换液无此净化效果。③通过简化实验步骤,减少取材接种时间,提高细胞活力。如胰酶消化后各个步骤均直接采用接种液PM为溶液,采用FBS中和胰酶。④增加离心后的过滤和沉淀。离心后还会有部分神经元缠结成絮状,无法吹匀,过滤弃除。仍有部分团块状的神经元得通过静置去除。
此外还有一些细节问题非常重要。吹打和消化是2个非常重要的因素。笔者的经验是:采用含EDTA的胰酶,利于吹散神经元;每次接种神经元之后15 min在光镜下观察神经元状态。如果团块细胞多,说明消化不够,需要增强消化时间或者胰酶浓度;如果有大量组织碎屑,说明消化过头;如果非神经元细胞过多,说明解剖不够干净准确。数次调整后摸索出适宜的消化方法和固定的吹打模式。不需要将所有的皮质组织都吹散利用,只需吹打出适当高活力的神经元即可。本组1只乳鼠的2个皮质可收集约300~400万个神经元。另外,接种密度也是重要的因素。接种密度过大会影响神经元生长,导致部分神经元死亡。接种的时候应该提前计算并混匀好接种液[8]。
本课题组培养的神经元生长发育较其它报道的模型快[9],1 d左右大多数神经元都长出突起,3 d左右神经元基本成熟,10 d左右出现衰老[10]。考虑可能和采用乳鼠皮质培养,较孕鼠神经元发育较快有关。另外,通过全量换液的过程,富集了高活力和生长发育快的神经元可能也是一个原因。
本研究通过简单的乳鼠皮质以及简便的培养过程,培养出高质量原代神经元,为类似的实验研究提供了参考,尤其是有关神经元死亡率相关的实验。
[1]Beaudoin GM 3rd,Lee SH,Singh D,et al.Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex[J].Nat Protoc,2012,7:1741-1754.
[2]Giordano G,Costa LG.Primary neurons in culture and neuronal cell lines for in vitro neurotoxicologicalstudies[J].Methods MolBiol,2011, 758:13-27.
[3]姜茜,姜玉武,王静敏,等.一种改进的大鼠皮层神经元原代培养方法及其性质鉴定[J].北京大学学报(医学版),2009,41:212-216.
[4]张小娟,李廷玉,刘友学,等.简易大鼠海马神经元原代培养方法及神经元兴奋性检测[J].南方医科大学学报,2010,30:2080-2083.
[5]康国创,刘文博,尹丽鹤,等.一种改进的高密度大鼠皮层神经元培养方法[J].中华神经外科疾病研究杂志,2009,8:417-420.
[6]何主强,杨国平,赵洪洋,等.Nogo-A受体在早期神经元细胞分化过程中的表达[J].神经损伤与功能重建,2015,9:95-97.
[7]戴锟,孙晓阳.成纤维细胞向神经元转化的研究进展[J].神经损伤与功能重建,2015,9:59-61.
[8]杨传豪,赵冬,刘祺,等.新生大鼠皮层神经元体外无血清原代培养[J].重庆医学,2014,43:3901-3903,3906.
[9]余华荣,杨贵忠,杨俊卿,等.大鼠皮层神经元原代培养方法的改进[J].重庆医科大学学报,2007,32:392-394,401.
[10]王旭辉,张岫竹,王伍超,等.大鼠下丘脑神经元培养的新方法[J].第三军医大学学报,2009,31:1709-1711.
(本文编辑:唐颖馨)
An Efficient and Convenient Model of Culturing Primary Neurons
ZHANGTao1、2,HUHuai-qiang1,WANGXu-hui3,NIUBing1,TAOZhen1,CAOBing-zhen1.
1.GeneralHospitalofJinanMilitaryArea,Jinan 250031,China;2.No.303Hospital ofChinesePeople'sLiberationArmy,Nanning530021,China;3.DapingHospital,ResearchInstituteofSurgeryThirdMilitaryMedical University,Chongqing400042,China
Objective:To establish an efficient and convenient model of culturing primary neurons.Methods: Firstly,the cortex of newborn Wistar mice younger than 12 hours old was thoroughly separated.And then the cortex was digested by 0.25%tyrisin for 30 min after cutting into pieces.Cells were rinsed,blew and centrifuged at 1000 rpm for 5 min after filtering.The supernatant was abandoned,and the resultant was added with plantation medium,gently blew and filtered.After standing for 3~5 min,the supernatant liquid was withdrawn in order to perform a count on the cells.The medium was replaced in full at 4 h,45 h and 96 h later after seeding and then the medium could be replaced in half every 3 days.At day 5 of culture,the neurons were evaluated by NSE,NMDAR1 and trypan blue staining.Results:The purity of neurons was over 95%and the mortality rate was less than 5%.The morphology of neurons was excellent,effectively crosslinking,and the background was clean.Con⁃clusion:Ahigh-quality,high-yield and simple neuronal culture model was established using neonatal rat cortex.
cortical neurons;primary culture;cell model;newborn rats
R741;R741.02
A DOI 10.16780/j.cnki.sjssgncj.2016.06.001
1.济南军区总医院济南 250031
2.解放军第303医院南宁 530021
3.第三军医大学大坪医院野战外科研究所重庆 400042
十二五全军重点项目No.BWS11J062
2016-01-20
曹秉振cbzxia2011@163. com
猜你喜欢
医院管理论坛(2022年9期)2022-10-28 05:51:28
北方药学(2022年3期)2022-09-06 00:22:06
中华实用诊断与治疗杂志(2022年1期)2022-08-31 09:58:22
解放军医学院学报(2020年12期)2020-03-29 05:11:32
中外医学研究(2019年27期)2019-04-21 13:35:19
电子测试(2018年13期)2018-09-26 03:29:26
福建畜牧兽医(2018年1期)2018-03-01 08:28:44
中西医结合心血管病电子杂志(2016年36期)2017-07-05 14:30:28
中国医学装备(2016年1期)2016-03-14 07:57:13
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:32
