
基于酯型生物碱含量变化选择蒸制附片
2016-04-08 08:37谭茂兰黄勤挽范润勇王智磊易佳佳
中成药 2016年2期
谭茂兰, 黄勤挽,2*, 肖 芳, 范润勇, 王智磊, 易佳佳
(1.成都中医药大学药学院,中药材标准化教育部重点实验室,四川省中药资源系统研究与开发利用重点实验室—省部共建国家重点实验室培育基地,四川成都611137;2.国家中医药管理局中药炮制技术重点研究室,四川成都611731)
基于酯型生物碱含量变化选择蒸制附片
谭茂兰1, 黄勤挽1,2*, 肖 芳1, 范润勇1, 王智磊1, 易佳佳1
(1.成都中医药大学药学院,中药材标准化教育部重点实验室,四川省中药资源系统研究与开发利用重点实验室—省部共建国家重点实验室培育基地,四川成都611137;2.国家中医药管理局中药炮制技术重点研究室,四川成都611731)
摘要:目的 基于6种酯型生物碱含有量的变化,选择合适的蒸制附片。方法 采用HPLC法,测定鲜附片、生附片、浸附片中3种双酯类生物碱(中乌头碱、乌头碱、次乌头碱)和3种单酯类生物碱(苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱)的含有量。结果 蒸制过程中双酯型生物碱的含有量迅速减少,而单酯型生物碱的含有量迅速增加,然后稳定在一定水平,但随蒸制时间延长会逐渐降低。结论 鲜附片更适合进行蒸制,蒸制时间以4~10 h为宜。
关键词:鲜附片;生附片;浸附片;蒸制;酯型生物碱;HPLC
dol:10.3969/j.issn.1001-1528.2016.02.029
KEY W 0RDS: fresh-cut FuPian;dried-cut FuPian;soaked-cut FuPian;steaming;ester-tyPe a1ka1oids;HPLC
附子为毛茛科植物乌头Aconitum carmichaeli Debx.的子根加工品,具有回阳救逆、补火助阳、散寒止痛功效,作为中药毒效鲜明的代表,被历代医家视为“回阳救逆第一要药”[1],因其含多种乌头碱类成分,毒性较强,故临床多用其炮制品,其加工过程主要为洗净、泡胆巴、煮、剥皮、切片、漂、蒸、晒。文献[2-6]表明,在传统加工过程中,较多水溶性成分(如毒效兼有的生物碱类)大量流失,导致药效药性受到一定影响,而且加工时胆巴的使用也在存争议。研究显示[7-8],附子的毒性成分主要为二萜双酯类生物碱,因其性质不稳定,在加热条件下易水解为低毒或无毒的产物(如单酯型、醇胺型生物碱),也可发生酯交换反应生成难溶于水的物质,从而减少毒性。为避免较多有效成分在加工过程中随水流失,故蒸制和炒制成为较受认同的附子炮制方法[9-10]。
目前,各类蒸附片并未对蒸制前附片的投料形式进行规定,也未见不同投料形式对蒸附片生物碱类成分含有量变化、成品性状差异等方面的相关报道[11-12]。本实验以不同状态的3种附片,即鲜切附片、鲜切70℃下干燥附片、水浸12 h生附片(简称鲜附片、生附片、浸附片)为对象,进行常压蒸制,根据《中国药典》2010年版附子含有量测定相关规定,分别测定3种双酯类毒性成分(中乌头碱、乌头碱、次乌头碱)和3种单酯类成分(苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱)在不同蒸制时间点的含有量,以研究不同状态附片在常压蒸制过程中6种酯型生物碱含有量的变化规律,初步探讨其在减毒存效方面的差异,确定适合常压蒸附片蒸制加工的投料形式和蒸制时间,完善相关炮制工艺。
1 仪器与试药
岛津LC-20A高效液相色谱仪,包括二元梯度泵、柱温箱、PDA检测器、自动进样器、Labso1ution工作站(日本岛津公司);BSA224S、BP211D电子分析天平(十万分之一,德国Sartorius公司);SHZ-DⅢ型循环水式多用真空泵(巩义市予华仪器有限责任公司);雷磁PHS-3C型PH计(上海仪电科学仪器股份有限公司);DHG-9240型电热恒温鼓风干燥箱(上海将任实验设备有限公司);FW135型中草药粉碎机(上海隆拓仪器设备有限公司);PS-80型超声波清洗机(深圳市洁康洗净电器有限公司);UPH-I-10T型优普超纯水器(成都超纯科技有限公司);RJTGL-16C型高速离心机(无锡瑞江分析仪器有限公司)。
泥附子(四川江油中坝附子科技发展有限公司),经成都中医药大学卢先明教授鉴定为毛茛科植物乌头Aconitum carmichaeli Debx.的子根,贮存于-20℃冰箱中备用。苯甲酰中乌头碱(批号120915)、苯甲酰乌头原碱(批号121109)、苯甲酰次乌头原碱(批号121209)、中乌头碱(批号121124)、乌头碱(批号121026)、次乌头碱(批号121218)对照品(成都普菲德生物技术有限公司,纯度均≥98%)。乙腈为色谱纯(美国Sigma-A1drich公司);氨水、异丙醇、乙酸乙酯、二氯甲烷、乙酸铵均为分析纯。
2 方法与结果
2.1 药材处理 取泥附子适量,解冻、洗净泥沙后纵切成片,厚度5 mm左右,均分为3份。取一份作为鲜附片,置于蒸锅上进行蒸制,圆气计时,保持锅内水沸腾和一定水量,分别于0、0.5、1、1.5、2、2.5、3、4、5、6、7、8、9、10、11、12 h取出适量,室温下放凉后于打粉机中混匀,冷藏备用。剩下两份于70℃烘箱中恒温干燥,干燥后取一份作为生附片,同法进行蒸制和样品处理;另一份参考江油中坝附子科技发展有限公司的蒸附片工艺,加入一定量水(没过药材表面)浸润12 h,沥去多余水分作为浸附片,同法进行蒸制和样品处理。
2.2 色谱条件 Phenomenex Gemini色谱柱(250 mm×4.6 mm,5 μm);柱温30℃;流动相A为0.04 mo1/L乙酸铵(氨水调PH至8.5),流动相B为乙腈,梯度洗脱(0~20 min,35%~40%B;20~25 min,40%~45%B;25~40 min,45%~50% B;40~50 min,50%~60% B);体积流量0.8 mL/min;检测波长235 nm;进样10 μL。对照品色谱图见图1,不同蒸制时间鲜附片样品色谱图见图2。
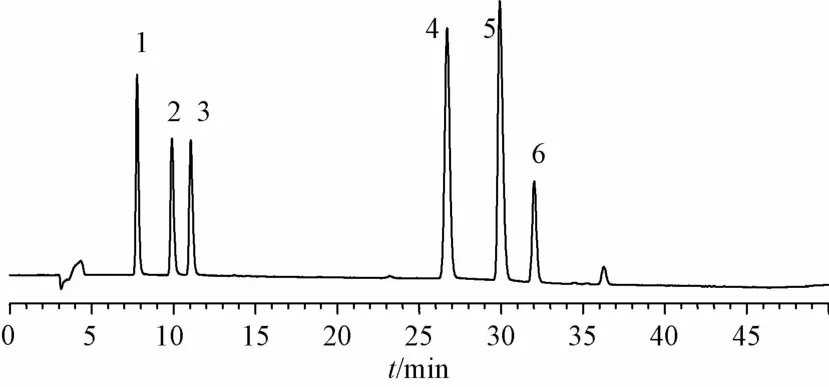
1.苯甲酰中乌头碱 2.苯甲酰乌头原碱 3.苯甲酰次乌头原碱4.中乌头碱 5.乌头碱 6.次乌头碱1.benzoy1mesaconitine 2.benzoy1aconine 3.benzoy1hyPaconine 4.mesaconitine 5.aconitine 6.hyPaconitine图1 6种酯型生物碱混合对照品的HP L C色谱图Flg.1 HPLC chromatograms of slx ester-type alkalold m lxed reference substances
2.3 对照品溶液的制备 分别精密称取苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、中乌头碱、乌头碱、次乌头碱对照品适量,0.05%盐酸-甲醇溶解,配制成每1 mL含苯甲酰中乌头碱0.484 mg、苯甲酰乌头原碱0.443 mg、苯甲酰次乌头原碱0.430 mg、中乌头碱0.398 mg、乌头碱0.345 mg、次乌头碱0.435 mg的原液。分别取苯甲酰中乌头碱1 mL、苯甲酰乌头原碱1 mL、苯甲酰次乌头原碱1 mL、中乌头碱3 mL、乌头碱1 mL、次乌头碱3 mL,置于同一10 mL量瓶中,0.05%盐酸-甲醇稀释至刻度,摇匀,得混合对照品溶液Ⅰ。精密吸取1 mL,置于10 mL量瓶中,0.05%盐酸-甲醇稀释至刻度,摇匀,得混合对照品Ⅱ。
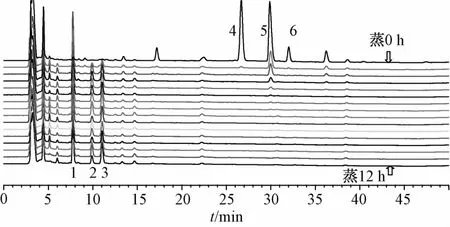
图2 不同蒸制时间鲜附片样品的H P L C色谱图(1~6同图1 )Flg.2 HPLC chromatograms of fresh-cut sllced aconlte sam ples ln dlfferent steam lng tlme (1~6 were same to Flg.1)
2.4 供试品溶液的制备[13]精密称取不同状态蒸制附片样品2 g,置于具塞锥形瓶中,加氨试液3 mL,精密加入异丙醇-乙酸乙酯(1∶1)混合溶液50 mL,称定质量,超声(300 W、40 kHz、25℃以下)30 min,放冷,再称定质量,异丙醇-乙酸乙酯(1∶1)混合溶液补足减失的质量,摇匀,滤过。精密量取续滤液25 mL,40℃以下减压回收溶剂至干,往残渣中精密加入0.05%盐酸-甲醇溶液3 mL溶解,16 000 r/min离心10 min,取适量上清液,即得。各样品平行制备2份。
2.5 供试品水分测定 按《中国药典》2010年版(一部)附录ⅨH水分测定法第一法(烘干法),测定各样品中的水分,平行测定2份。
2.6 方法学考察结果
2.6.1 线性关系考察 精密吸取混合对照品溶液Ⅱ1、5、10 μL和Ⅰ5、10、20、40 μL,分别注入液相色谱仪,在“2.2”项色谱条件下进行测定,以进样量(μg)为横坐标(X),峰面积(A)为纵坐标(Y)绘制标准曲线,结果见表1。

表1 标准曲线方程及线性关系Tab.1 Standard curve equatlons and llnear relatlonsh lps
2.6.2 精密度试验 精密吸取混合对照品溶液Ⅰ10 μL,在“2.2”项色谱条件下重复进样6次,测定6种酯型生物碱的峰面积。结果,苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、中乌头碱、乌头碱、次乌头碱的峰面积RSD分别为0.83%、1.1%、1.2%、1.0%、0.86%、1.3%,表明仪器精密度良好。
2.6.3 稳定性试验 取鲜附片蒸制0.5 h后的样品1份,按“2.4”项下方法制备供试品溶液,在“2.2”项色谱条件下,分别于0、4、8、12、24 h进样测定,记录6种酯型生物碱的峰面积。结果,苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、中乌头碱、乌头碱、次乌头碱的峰面积RSD分别为1.0%、1.2%、1.1%、1.4%、 1.2%、1.8%,表明溶液在24 h内稳定。
2.6.4 重复性试验 取鲜附片蒸制0.5 h后的样品6份,按“2.4”项下方法制备供试品溶液,在“2.2”项色谱条件下测定,计算6种酯型生物碱的含有量。结果,苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、中乌头碱、乌头碱、次乌头碱含有量的RSD分别为2.7%、2.2%、2.6%、2.2%、2.6%、1.8%,表明该方法重复性良好。
2.6.5 加样回收试验 取含有量已知的鲜附片蒸制0.5 h后的样品6份,分别精密加入一定量的对照品原液(苯甲酰中乌头碱0.6 mL、苯甲酰乌头原碱0.1 mL、苯甲酰次乌头原碱0.15 mL、中乌头碱0.05 mL、乌头碱0.2 mL、次乌头碱0.05 mL),按“2.4”项下方法制备供试品溶液,在“2.2”项色谱条件下进样5 μL,测定6种酯型生物碱的含有量,计算平均回收率和RSD,结果见表2。
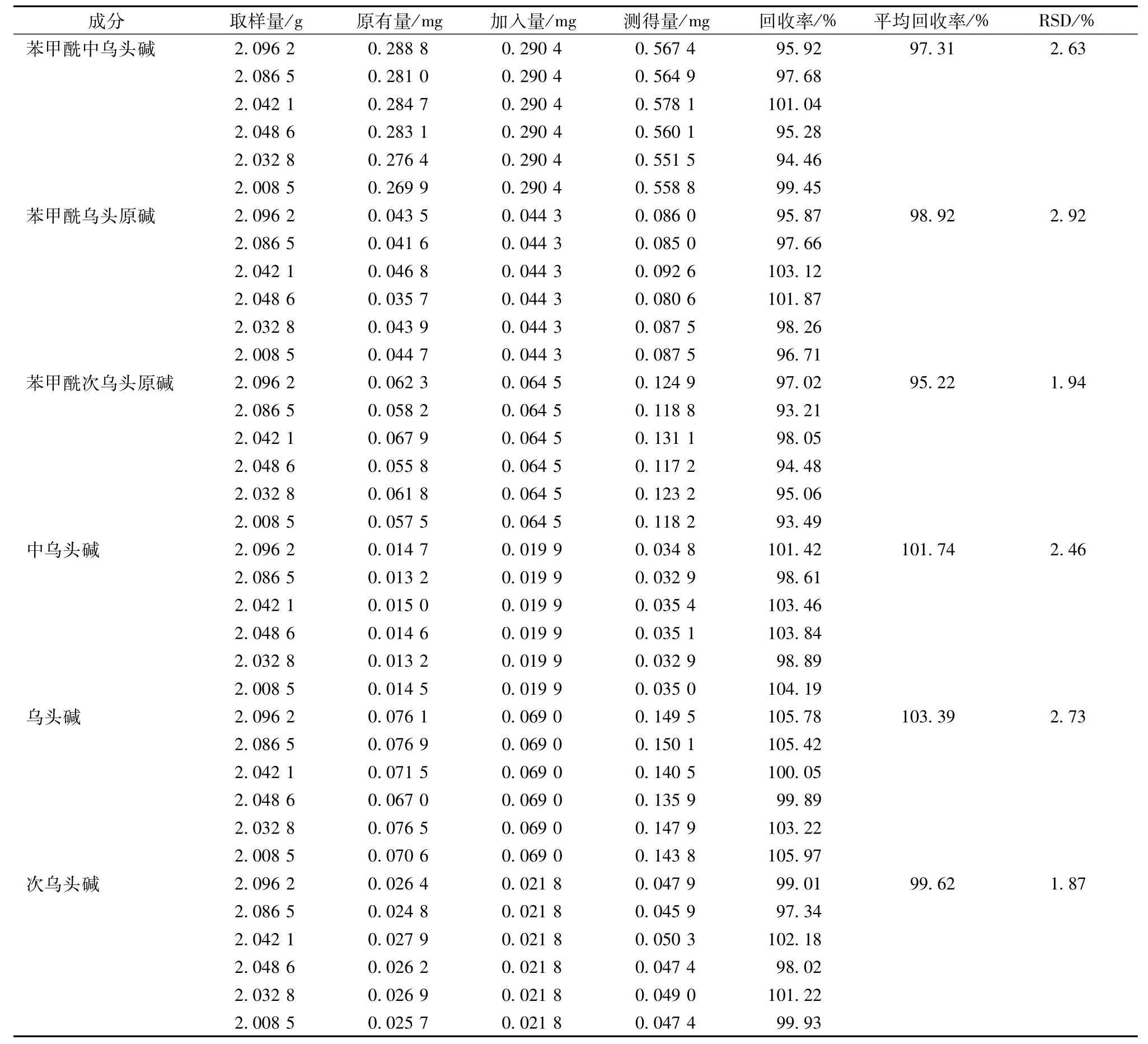
表2 加样回收率试验结果Tab.2 Results of recovery tests
2.7 供试品含有量测定 在“2.2”项色谱条件下,测定苯甲酰中乌头碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、中乌头碱、乌头碱、次乌头碱的峰面积,以标准曲线法计算含有量。按干燥品计,3种状态附片的含有量测定结果分别见表3~5(各表中数据均由标准曲线线性范围内的原始数据计算得出)。

表3 不同蒸制时间鲜附片中6种酯型生物碱的含有量(m g/g,n=2)Tab.3 Contents of slx ester-type alkalolds ln fresh-cut sllced aconlte ln dlfferent steam lng tlme(mg/g,n=2)
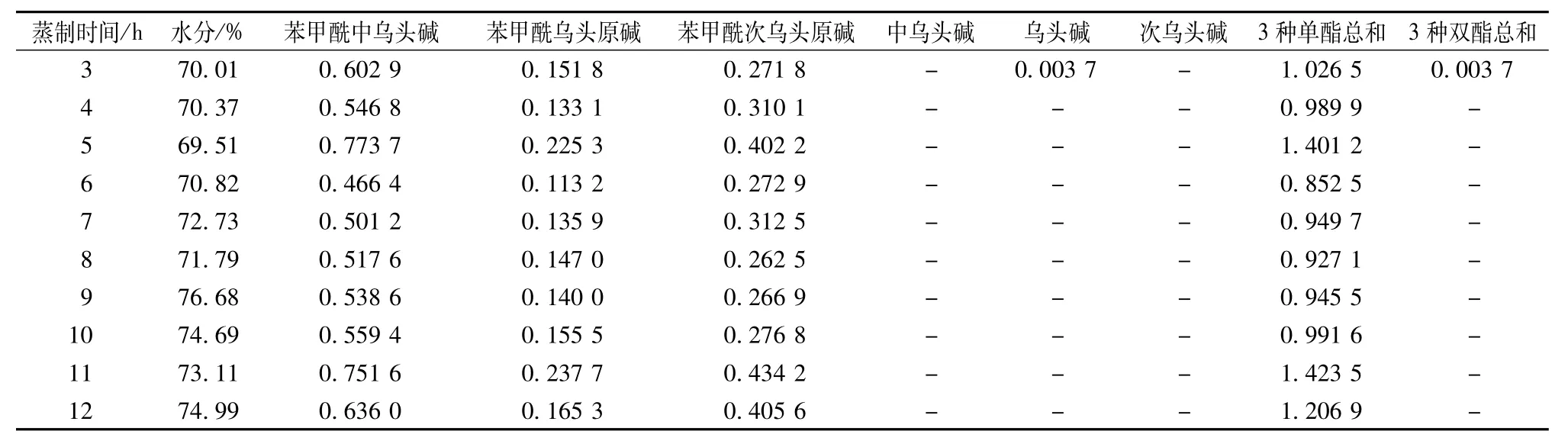
续表3

表4 不同蒸制时间生附片中6种酯型生物碱的含有量(m g/g,n=2)Tab.4 Contents of slx ester-type alkalolds ln drled sllced aconlte ln d lfferent steam lng tlme(mg/g,n=2)
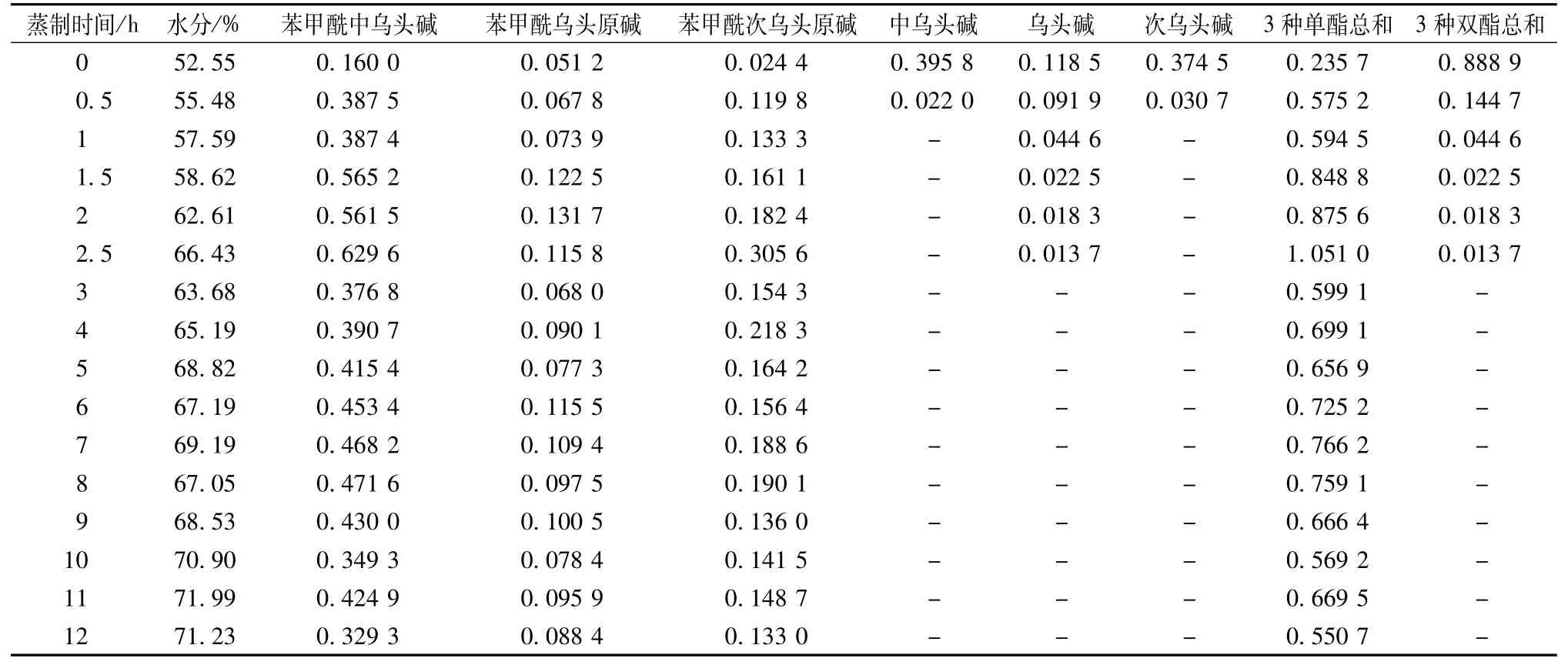
表5 不同蒸制时间浸附片中6种酯型生物碱的含有量(m g/g,n=2)Tab.5 Contents of slx ester-type alkalolds ln soaked sllced aconlte ln d lfferent steam lng tlme(mg/g,n=2)
3种单酯、3种双酯的含有量总和见图3、图4。样品的提取方法参照现行版药典,色谱条件根据文献[14]进行部分调整,考察了流动相A在不调PH和PH分别为8.5、9、10条件下对6种酯型生物碱分离效果的影响。考虑到样品数量和乌头碱的毒性,将流动相A调PH至8.5,此时其分离度较佳,分析时间较短。
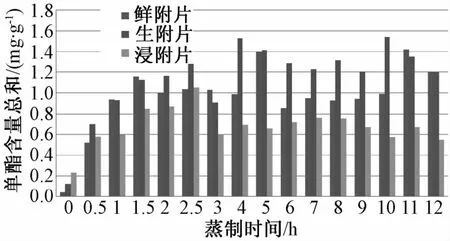
图3 3种单酯含有量总和的柱状图Flg.3 H lstogram s of the total contents of three monoester alkalolds

图4 3种双酯含有量总和的柱状图Flg.4 H lstograms of the total contents of three dlester alkalolds
由图4可知,各状态附片在蒸制过程中,双酯型生物碱含有量的变化相似,均在蒸制0.5 h内显著降低,然后逐渐减少至无法检出。由图3可知,单酯型生物碱含有量的变化较复杂,不同状态附片之间存在一定差异,结合总体变化趋势及双酯型生物碱含有量变化,可将各状态附片中3种单酯型生物碱含有量总和的变化分为3个阶段:(1)转化上升阶段。单酯型生物碱含有量随双酯型生物碱的降低而逐渐升高,总和呈增长趋势。鲜附片蒸制转化的上升期为0~1.5 h,生附片和浸附片为0~2.5 h;(2)叠加反应阶段。各状态附片单酯型生物碱的含有量均继上升期降低,再经一段波动达一最大值后,稳定在一定范围内。鲜附片蒸制的叠加反应期在1.5~10 h之间,蒸制5 h达到最高;生附片在2.5~9 h之间,蒸制4 h达到最高;浸附片在2.5~10 h之间,蒸制7 h达到最高。此阶段单酯型生物碱含有量的波动参考文献[15],推断可能与其水解、酯交换[7]、双酯型生物碱的水解等反应相关,是多个反应叠加的结果;(3)溶胀波动阶段。各状态附片中单酯型生物碱的含有量均出现先升后降的变化,鲜附片和浸附片蒸制的溶胀波动期是10~12 h,10~11 h含有量总和升高,11~12 h降低至蒸制结束;生附片是9~12 h,9~10 h含有量升高,然后降低至蒸制结束。此阶段的波动原因除与酯型生物碱之间的转化有关外,还可能与此时附片的形态有关。经长时间蒸制后,各附片含水量均达到70%左右,表面均有较多的淀粉溶胀,推断此时附片组织、细胞间的连接已被破坏,细胞通透性的增加有利于生物碱的溶出,而且各成分随水汽损失的量亦有所增加,导致出现上述波动。单个生物碱含有量的变化规律与其总和类似,在此不再赘述。
另外,本实验选择以蒸制后的样品直接混匀制样,未经干燥,减少了其他因素对生物碱含有量的影响,更能真实反映蒸制过程中6种生物碱的含量变化。
3 讨论与结论
3种不同状态附片的主要差异为初始含水量和组织间的紧密度。其中,鲜附片是附子的原本状态;生附片含水量低,组织间紧密度高;浸附片含水量和组间紧密度则介于鲜附片和生附片之间,是人为处理附片最常见的状态。在相同的蒸制条件下,对湿热敏感的双酯型生物碱反应接近,不同状态附片均在蒸制1 h后,双酯型生物碱的含有量达到现行版药典要求(总和不得超过0.02%),虽然其减毒程度相近,但存效程度存在差异,鲜附片和浸附片双酯型生物碱在蒸制3 h后均低于检测限,而生附片则在11 h内持续检出,说明附片蒸制时其减毒程度与物料的初始含水量和质地相关,保持一定初始含水量可增加附子的减毒程度。
单酯型生物碱作为双酯型生物碱转化的产物,其含有量的高低可说明附子解毒的程度,因其对热稳定,故也可反映附片成分的保留情况。3种附片中单酯型生物碱的含有量均在蒸制0.5 h后达到药典要求(总和不得少于0.01%),说明减毒程度相当。各附片蒸制12 h后的含有量仍合格,说明蒸制能较好地保留附子成分,保证附子药效。在蒸制过程中,生附片单酯型生物碱的含有量高于鲜附片,说明初始含水量越低,质地越紧密,单酯含有量保存越好,成分损失越少。
实验显示,生附片直接蒸制时因其质地紧密,导致水蒸汽穿透、渗入缓慢,其成分损失较少,单酯含有量保存较好,但其减毒程度也较低。而且,蒸制时其干湿程度不均,导致淀粉类成分溶胀也不均,附片呈稀糊状,颜色偏深,并难以保持片型,所以常压蒸附时不宜用生附片直接蒸制;鲜附片双酯转化快,转化程度高,加工工序简单,单酯含有量高,变化较稳定,样品的颜色和片型均较理想。样品蒸制6 h干燥后,呈透明黄棕色角质样,与黑顺片类似,故常压蒸制附片时建议以鲜附片进行投料;考虑到附子的保存和大量加工的需要,大生产可以浸附片投料蒸制,因其蒸制后片型也较好,蒸制6 h干燥后呈半透明或不透明黑棕色角质样,与黑顺片差异较大,但各生物碱含有量仍满足药典规定,只是加工工序较多、时间较长。
另外,浸附片存在浸润时成分随水分流失的情况,其单酯含有量总体水平低于鲜附片和生附片,所以应严格控制浸润水量和时间。查阅相关资料发现,对生附片浸润水量并未有明确的规定和研究,故本实验在参考文献[16]的基础上,对生附片吸水量作了简单考察,发现浸润12 h时生附片的吸水量为1∶1,故建议浸润加水量在1∶1~1∶1.5之间。以单酯型生物碱的含有量为指标,初步确定常压蒸附片蒸制时间以4~10 h为宜,但最佳蒸制时间点还需深入考察。另外,以鲜附片和浸附片投料制备的蒸附片性状的差异,药效、毒性、药性等方面是否与黑顺片相近等问题也需进行进一步研究。
参考文献:
[1] 张世臣,李 可.中国附子[M].北京:中国中医药出版社,2013: 8.
[2] 刘鸿鸣,赵幼祥.附子炮炙研究[M].成都:四川省中药研究所,1958.
[3] 郑露露.附子炮制中的成分流失[J].中药通报,1983,8 (2): 26-28.
[4] 叶 强,郭一平,彭 成.炮制方法对附子生物碱类成分的影响[J].华西药学杂志,2013,28(3): 275-277.
[5] 周 林,李 飞,任玉珍,等.附子中生物碱含量与在胆巴液中浸泡时间变化规律的研究[J].中国实验方剂学杂志,2014,20(10): 71-74.
[6] 周远鹏.附子研究的回顾和思考-生附子去毒和心血管活性部位或成分的研究(三)[J].中药药理与临床,2014,30(4): 131-134.
[7] 周远鹏.从双酯型生物碱的水解来看附子的解毒(二)[J].中药药理与临床,2014,30(3): 154-157.
[8] Wang Y,Shi L,Song F R,et al.ExP1oring the ester-exchange reactions of dister-diterPenoid a1ka1oids in the aconite decoction Process by e1ectrosPray ionization tandem mass sPectometry[J].RaPid Commun Mass SPectrom,2003,17(4): 279-284.
[9] 温瑞卿,李东辉,赵 昕,等.基于化学分析的毒性中药附子炮制方法的合理性研究[J].药学学报,2013,48 (2): 286-290.
[10] 匡青芬,侯大斌,孙 鸿,等.附子无胆炮制品的生物碱与可溶性多糖含量检定[J].时珍国医国药,2014,25 (4): 850-852.
[11] 朱日然,李启燕,朱宗敏,等.HPLC法测定附子与其炮制品中双酯型生物碱[J].中成药,2011,33(8): 1375-1378.
[12] 方 莉,林 华,邓广海,等.正交试验法优选附子高压蒸制工艺[J].中国实验方剂学杂志,2012,18(23): 20-24.
[13] 国家药典委员会.中华人民共和国药典: 2010年版一部[S].北京:中国医药科技出版社,2010: 177.
[14] 周子渝.基于附子“成分-药材-饮片-复方”毒性变化规律的研究[D].成都:成都中医药大学,2012.
[15] 郑 琴,陆浩伟,郝伟伟,等.乌头类双酯型生物碱水解转化规律及含量计算方法研究[J].中国药学杂志,2011,46(9): 652-655.
[16] 陈彦琳.附子加压蒸制工艺及其饮片的适用性研究[D].北京:北京中医药大学,2007.
Selectlon of sultable herbs for steam lng Fuplan based on the change of contents of ester-type alkalolds
TAN Mao-1an1, HUANG Qin-wan1,2*, XIAO Fang1, FAN Run-yong1, WANG Zhi-1ei1, YI Jia-jia1
(1.Collegeof Pharmacy,Chengdu University of TraditionalChineseMedicine;Key Laboratory of Standardization of ChineseHerbalMedicine,Ministry of Education;Sichuan Provincial Key Laboratory of Systematic Research,DeveloPmentand Utilization of ChineseMedicine Resources—Key Laboratory Breeding Baseof Co-founded by Sichuan Province and MOST,Chengdu 611137,China;2.Key Laboratory of Technology of ChineseMedicine Processing,State Administration of Traditional ChineseMedicine,Chengdu 611731,China)
ABSTRACT:AIM To se1ect the suitab1e herbs for steaming FuPian(Radix Aconiti 1atera1is PreParata s1ice)based on the change of contents of six ester-tyPe a1ka1oids.METH0DS HPLC was used for determining the contents of three diester-tyPe a1ka1oids(mesaconitine,aconitine,hyPaconitine)and three monoester-tyPe a1ka1oids (benzoy1mesaconitine,benzoy1aconine,benzoy1hyPaconine)in fresh,dried and soaked-cut FuPian.RESULTS The contents of diester-tyPe a1ka1oidswere decreased significant1y,whi1e those ofmonoester-tyPe a1ka1oidswere increased raPid1y and then stabi1ized at a certain 1eve1.Butwith the extension of steaming time,the contents ofmonoester-tyPe a1ka1oids were a1so decreased gradua11y.C0NCLUSI0N Fresh-cut FuPian is more suitab1e for steaming,and the steaming time is 4 -10 hours.
*通信作者:黄勤挽(1979—),男,副教授,硕士生导师,研究方向为中药炮制与制剂。Te1:(028)61801001,E-mai1: 36190587@ qq.com
作者简介:谭茂兰(1990—),女,硕士,研究方向为中药炮制与制剂。Te1: 15281090450,E-mai1: tanmao1an2010@163.com
基金项目:国家自然科学基金(81274086);四川省科技厅支撑计划(2013SZ0140);四川省中医药管理局青年基金(2010-12)
收稿日期:2015-08-03
中图分类号:R283
文献标志码:A
文章编号:1001-1528(2016)02-0366-07
