
胆汁淤积性肝病诊断和治疗共识(2015)*
2016-04-07 02:23中华医学会肝病学分会,中华医学会消化病学分会,中华医学会感染病学分会
胃肠病学 2016年1期
胆汁淤积性肝病诊断和治疗共识(2015)*
中华医学会肝病学分会中华医学会消化病学分会中华医学会感染病学分会
关键词胆汁淤积性肝病;诊断;治疗;共识
Consensus on the Diagnosis and Treatment of Cholestatic Liver Diseases
ChineseSocietyofHepatology,ChineseMedicalAssociation;ChineseSocietyofGastroenterology,ChineseMedicalAssociation;ChineseSocietyofInfectiousDiseases,ChineseMedicalAssociation.
Key wordsCholestatic Liver Diseases;Diagnosis;Treatment;Consensus
一、概述
胆汁淤积(cholestasis)是指肝内外各种原因造成胆汁形成、分泌和排泄障碍,胆汁流不能正常流入十二指肠而进入血液的病理状态,临床可表现为瘙痒、疲劳、尿色加深和黄疸等,早期常无症状,仅表现为血清碱性磷酸酶(alkaline phosphatase, ALP)和γ-谷氨酰转肽酶(gamma-glutamyl transferase, GGT)水平升高,病情进展后可出现高胆红素血症,严重者可导致肝功能衰竭甚至死亡[1]。以各种原因使肝脏病变导致胆汁淤积为主要表现的肝胆疾病统称为胆汁淤积性肝病,胆汁淤积本身也会进一步加重肝脏的损害。胆汁淤积性肝病根据发生部位可分为肝内胆汁淤积和肝外胆汁淤积。如胆汁淤积持续超过6个月,则称为慢性胆汁淤积[2]。本共识主要介绍肝内胆汁淤积性肝病。
为帮助我国临床医师规范诊治胆汁淤积性肝病和开展相关科研工作,中华医学会肝病学分会、中华医学会消化病学分会和中华医学会感染病学分会组织国内有关专家对胆汁淤积性肝病的定义、发生机制、临床表现、诊断标准和治疗原则以及相关疾病进行了较为全面的描述,并根据循证医学原则提出建议,形成了《胆汁淤积性肝病诊断和治疗共识》一文。随着我国胆汁淤积性肝病临床资料的不断积累和完善,本共识将适时更新。
本共识采用推荐分级的评估、制定和评价(GRADE)系统对循证医学证据的质量(表1)和推荐意见的级别(表2)进行评估[3]。在形成推荐意见时,不仅考虑到证据的质量,还应权衡干预的利弊与负担、患者偏好和价值观的可变性以及资源的合理利用、推荐措施的公平性和可实施性等因素。

表1 GRADE 系统证据质量及其定义

表2 GRADE系统推荐强度等级
二、流行病学
胆汁淤积性肝病的发生率目前尚无确切的数据,主要因其诊断标准未能统一,以致结果不甚一致。1992年Bortolini等[4]对初次诊断慢性肝病患者胆汁淤积的流行病学情况进行了多中心调查,胆汁淤积的诊断标准为在排除机械性胆管梗阻和胆管手术史的情况下,血清总胆红素>18 μmol/L、直接胆红素>4 μmol/L和ALP>2 μkat/L(kat 是酶的国际活性单位)。结果显示在2 520例初次诊断慢性肝病患者中,882例(35.0%)出现胆汁淤积。胆汁淤积更易出现在原发性胆汁性肝硬化(primary biliary cirrhosis, PBC)/原发性胆汁性胆管炎(primary biliary cholangitis)、原发性硬化性胆管炎(primary sclerosing cholangitis,PSC)中,并发现黄疸较瘙痒在伴有胆汁淤积的患者中更常见。我国1 000 例病毒性肝炎患者胆汁淤积的横断面研究[5]显示,56%慢性病毒性肝炎患者出院时,ALP 或GGT 仍高于正常值上限(ULN),且在这些指标异常的患者中肝纤维化和肝硬化的发生风险和病情严重程度显著增加。曹旬旬等[6]以ALP水平高于1.5倍ULN,且GGT水平高于3倍ULN为诊断标准,对4 660例上海市住院慢性肝病患者胆汁淤积发生情况进行研究,结果显示总发生率为10.26%,慢性肝病患者胆汁淤积发生率随年龄增加呈上升趋势。在不同慢性肝病中,胆汁淤积发生率较高的为PSC(75.00%)、各种原因肝硬化(47.76%)和PBC(42.86%),其他依次为肝肿瘤(35.97%)、自身免疫性肝炎(AIH)(30.77%)、药物性肝病(28.31%)、酒精性肝炎(16.46%)、病毒性肝炎(5.22%)和非酒精性脂肪性肝病(NAFLD)(2.70%)。
三、病因和分类
引起胆汁淤积的原因较多,常见病因主要有病毒、细菌、寄生虫、药物和(或)毒物、自身免疫、乙醇、结石、肿瘤和遗传代谢等,任何能引起肝细胞和胆管细胞损害以及胆管系统梗阻的因素均可导致胆汁淤积的发生。肝细胞功能障碍或毛细胆管、细胆管(<15 μm,亦称闰管或Hering 管)和小叶间胆管(15~100 μm)病变或阻塞所致胆汁淤积称为肝内胆汁淤积[2,7-8];间隔胆管(>100 μm)、区域胆管(300~400 μm)、节段胆管(400~800 μm)、左右肝管、胆总管至壶腹部的病变或阻塞所致胆汁淤积称为肝外胆汁淤积。大多数胆汁淤积性肝病是肝内胆汁淤积,而PSC 可累及小和大肝内胆管和(或)肝外胆管,因此部分患者可同时有肝内和肝外部分病变[2,8]。
1. 肝内胆汁淤积:根据细胞学损害的部位可分为肝细胞性和胆管细胞性[2]:①肝细胞性胆汁淤积主要病因有败血症和毒血症、病毒性肝炎、酒精或非酒精性脂肪性肝炎(NASH)、药物或胃肠外营养、遗传性疾病[如良性复发性肝内胆汁淤积(benign recurrent intrahepatic cholestasis, BRIC)、进行性家族性肝内胆汁淤积(progressive familial intrahepatic cholestasis, PFIC)]、妊娠期肝内胆汁淤积(intrahepatic cholestasis of pregnancy, ICP)、红细胞生成性原卟啉症、恶性浸润性疾病(如造血系统的霍奇金病和转移性肿瘤)、良性浸润性疾病(如淀粉样变性、肉芽肿性肝炎和肉芽肿病)、管壁发育异常(如先天性肝纤维化)、血管性疾病(如Budd-Chiari综合征和静脉闭塞性疾病)、肝硬化(各种原因)。②胆管细胞性胆汁淤积主要疾病和病因有PBC、PSC以及合并AIH重叠综合征、特发性成人肝内胆管缺失症、管壁发育异常(如胆汁性错构瘤和Caroli综合征)、囊性纤维化、药物性胆管病、移植物抗宿主病和继发性硬化性胆管炎,后者包括各种胆石病、缺血性胆管病(遗传性出血性毛细血管扩张症、结节性多动脉炎和其他类型的脉管炎)、获得性免疫缺陷综合征和其他类型的免疫抑制相关的感染性胆管炎等。肝细胞和胆管细胞均有损害者称为混合性胆汁淤积。
2. 肝外胆汁淤积:主要疾病和病因有PSC、胆管结石、先天性肝外胆管闭锁、胆总管/Oddi 括约肌狭窄、胆管寄生虫病、胆总管囊肿、肿瘤性疾病(胆总管癌、肝细胞癌侵及胆管、壶腹部癌、胆总管旁淋巴结转移压迫)、胰腺疾病(胰腺癌、胰腺囊肿和慢性胰腺炎)等。
四、临床表现和实验室检查
除引起胆汁淤积原发疾病的相关临床症状外,肝脏胆汁淤积本身可引起相关临床症状以及因胆汁淤积而致的继发性改变。患者早期可无不适症状,可有疲劳、纳差、恶心、上腹不适等非特异性症状,胆汁淤积相关的临床表现主要有黄疸、皮肤瘙痒、疲劳、脂肪泻、黄色瘤和骨质疏松等。胆汁淤积引起的黄疸以直接胆红素升高为主,肝细胞损害引起的黄疸因同时有摄取、结合、排泄的障碍,因此直接和间接胆红素均可升高,但一般直接胆红素升高的幅度较间接胆红素大。血清ALP和GGT升高是胆汁淤积最具有特征性的早期表现,两者升高提示出现胆汁淤积。ALP和GGT均表达于肝细胞血窦侧和毛细胆管侧以及胆管细胞微绒毛上,经胆汁排入胆管系统。当胆汁排泄不畅时,毛细胆管内压增高,可诱发ALP产生增多,加之胆汁酸凭借其表面活性作用,将ALP从脂质膜上溶析出来,使血清ALP明显增高[1,9]。除肝内外胆汁淤积相关疾病外,妊娠、儿童生长期、骨骼疾病以及部分肿瘤亦可出现ALP升高。GGT增高较其他血清酶出现得更早,持续时间更长,在肝酶中敏感性最高,但特异性较低。血清GGT对胆汁淤积的诊断敏感性和特异性可能不低于甚至优于ALP。在排除酗酒等其他肝损伤因素的情况下,若ALP和GGT同时升高,可确认存在肝细胞和胆管细胞损伤。若GGT升高而ALP正常,几乎也可判定存在肝毛细胆管和胆管上皮细胞损伤。
若GGT正常而ALP升高,则应考虑骨病等可能。在ALP升高的病例中,如不合并GGT升高,常可排除肝源性疾病。需注意的是在一些特殊胆汁淤积性肝病如PFIC 1型和2型以及BRIC等,GGT可不增高。胆汁酸在肝内合成、分泌,其血清水平升高是胆汁淤积敏感和早期特异性指标。正常人肝脏合成的胆汁酸包括胆酸(cholic acid, CA)、鹅去氧胆酸(chenodeoxycholic acid, CDCA)和代谢中产生的脱氧胆酸(deoxycholic acid, DCA),还有少量石胆酸(lithocholic acid, LCA)和微量熊去氧胆酸(ursodeoxycholic acid, UDCA),合称总胆汁酸(total bile acid, TBA)。血清胆汁酸的定量测定可作为检测胆汁淤积的一种敏感且特异的方法。发生胆汁淤积时,胆汁分泌下降,并迅速改变胆汁酸贮存量的分布,使血清和尿液中的胆汁酸浓度显著升高。血清胆汁酸对诊断胆汁分泌受损较血清胆红素更为敏感,但对大多数胆汁淤积的敏感性不如ALP,且许多肝病如肝硬化、急慢性肝炎均可有胆汁酸升高。正常空腹胆汁酸为1.0~6.0 μmol/L,餐后2 h为 6.0~9.0 μmol/L。胆汁淤积时胆汁酸超过10 μmol/L。胆汁酸10~20 μmol/L为轻度增高,20~40 μmol/L为中度增高,40 μmol/L以上为重度增高[1]。此外,甘胆酸(一种甘氨酸结合型胆汁酸)检测已进入临床应用阶段,动态观察有益于临床发现胆汁淤积,特别是对ICP的判断具有重要的临床意义,但由于目前检测方法缺乏标准致其临床价值受限。胆汁酸和甘胆酸虽然均是反映胆汁淤积的敏感指标,但检测方法缺乏标准,加上干扰因素多、特异性欠佳等因素,故目前国内外相关指南中未将其列入检测方法并细化判断标准。胆汁淤积时丙氨酸氨基转移酶(ALT)和天冬氨酸氨基转移酶(AST)一般不高,仅当胆汁淤积引起肝细胞损害时才会出现ALT和AST升高,可伴有血清胆固醇、磷脂、三酰甘油均升高,血清脂蛋白异常。检测血清中自身抗体如抗核抗体(ANA)、抗平滑肌抗体(SMA)、抗肝肾微粒体抗体(LKM)、抗肝细胞胞浆抗原1 型抗体(LC-1)、抗线粒体抗体(AMA)、抗Sp100抗体、抗可溶性肝抗原抗体(SLA/LP)等有助于进一步明确胆汁淤积的病因。
五、影像学和内镜检查
腹部超声检查通常用于了解肝内外胆管是否阻塞扩张。腹部CT检查对胆管梗阻性病变的诊断有一定的价值。磁共振胰胆管造影(magnetic resonance cholangiopancreatography, MRCP)是胆管系统安全而又准确的检查,对胆管系统梗阻诊断的准确性接近内镜逆行胰胆管造影(endoscopic retrograde cholangiopancreatography, ERCP)。内镜超声(endoscopic ultrasonography, EUS)检查在检测胆管结石以及引起肝外胆管梗阻的病变方面的准确性与MRCP相当。诊断和治疗肝外胆管梗阻的金标准为ERCP,但其具有创伤性。即使是经验丰富的操作者仍可导致较多的并发症,如手术相关胰腺炎、出血和胆管炎等。因此,在考虑肝外胆管梗阻且尚不确定是否需要内镜干预时,应首先行MRCP或EUS,以避免不必要的ERCP。如上述检查不能明确诊断,需行肝活组织病理学检查以进一步明确诊断。
六、组织病理学检查
胆汁淤积的大体标本呈黄绿色,穿刺标本呈散在绿色斑点或通体深绿色。肝内胆汁淤积的组织病理特征是胆汁从肝小叶第三区肝细胞开始,表现为肝细胞内胆汁淤积,肝细胞呈羽毛状变性,伴毛细胆管扩张,胆栓形成。严重时以扩张含胆栓的毛细胆管为中心,肝细胞呈腺泡样排列,形成胆汁花环。肝窦内增生肥大的Kupffer细胞吞噬胆汁,门管区小叶间胆管胆汁淤积伴胆栓形成。电镜观察显示毛细胆管微绒毛水肿、变短,直至消失。肝外阻塞性胆汁淤积的组织病理学特征为门管区周边肝内胆汁湖伴胆汁肉芽肿形成,长期肝外阻塞可引起肝内继发性胆汁淤积。胆汁淤积后期可引起门管区纤维化,甚至胆汁性肝硬化[7,10]。
七、诊断和鉴别诊断
1. 胆汁淤积性肝病诊断标准:目前尚无统一的诊断标准,以ALP和GGT作为诊断指标尚有一些争议。2009年欧洲肝病学会(EASL)《胆汁淤积性肝病的处理临床实践指南》[2]建议:ALP超过1.5倍ULN且GGT超过3倍ULN可诊断胆汁淤积性肝病。鉴于我国现状以及有利于国际交流,本共识仍推荐2009年EASL的指南作为胆汁淤积诊断标准。但需注意一些特殊胆汁淤积性肝病,如PFIC 1型和2型以及BRIC等,GGT可不升高。
2. 诊断步骤:诊断胆汁淤积性肝病分三个步骤。首先,确定胆汁淤积是否存在,可通过血清学方法确定;其次,影像学和内镜检查确定是阻塞性还是非阻塞性;第三,综合分析得出诊断[包括病因、肝组织病理学、ERCP和经皮肝穿刺胆管造影(percutaneous transhepatic cholangiography, PTC)以及基因检测等]。仔细询问病史和体格检查对诊断很重要,包括职业、药物史、饮酒史和家族史等。部分胆汁淤积性肝病仅见于某些特殊情况,如妊娠、儿童、肝移植、人类免疫缺陷病毒感染。腹部超声检查可了解有无肝内外胆管扩张。胆总管扩张且内径超过8 mm高度提示肝外梗阻。MRCP是检查胆管系统的安全又准确的方法。ERCP是显示胆管和治疗肝外胆管梗阻的金标准,但即使是有经验的操作者,仍有较高的并发症发生率(3%~5%发生胰腺炎;行括约肌切开术时,2%合并出血,1%合并胆管炎,0.4%发生操作相关的死亡)[11]。因此,在考虑肝外胆管梗阻且尚不确定是否需行内镜干预时,应首先行MRCP或EUS,然后再考虑ERCP[12]。排除肝外梗阻后,进一步的检查是检测血清肝炎病毒标记物、肝病相关的自身抗体如AMA。高滴度AMA(≥1/40)且ALP很高,并在缺乏其他解释时,可诊断为PBC。对原因不明的胆汁淤积性肝病患者,如AMA阴性,下一步可行MRCP,必要时行ERCP。如诊断仍不明确,可行肝组织活检。行组织学评估时,应特别注意胆管病变;对于AMA 阴性和肝组织活检未能确诊的患者,有条件者可考虑基因检测,如ABCB4等基因。胆汁淤积性肝病诊断流程见图1[2]。
3. 与黄疸的鉴别和联系:胆汁淤积与黄疸不完全等同,胆汁淤积是包括胆红素在内的全部胆汁成分淤积[1]。黄疸是血液胆红素浓度增高,使巩膜、皮肤等组织发生黄染的现象。有些疾病仅有胆红素代谢障碍,而胆汁其他成分分泌和排泄正常,如遗传性高胆红素血症(Gilbert 综合征、Crigler-Najjar综合征、Dubin-Johnson综合征和Rotor综合征等)和溶血性疾病,这些患者仅有胆红素升高,而ALP、GGT和胆汁酸并不升高。胆汁淤积早期,仅有ALP、GGT和胆汁酸升高,不一定出现黄疸,通常仅当胆红素超过34.2 μmol/L时临床上才显现黄疸。因此黄疸患者需排除遗传性高胆红素血症和血液系统疾病。
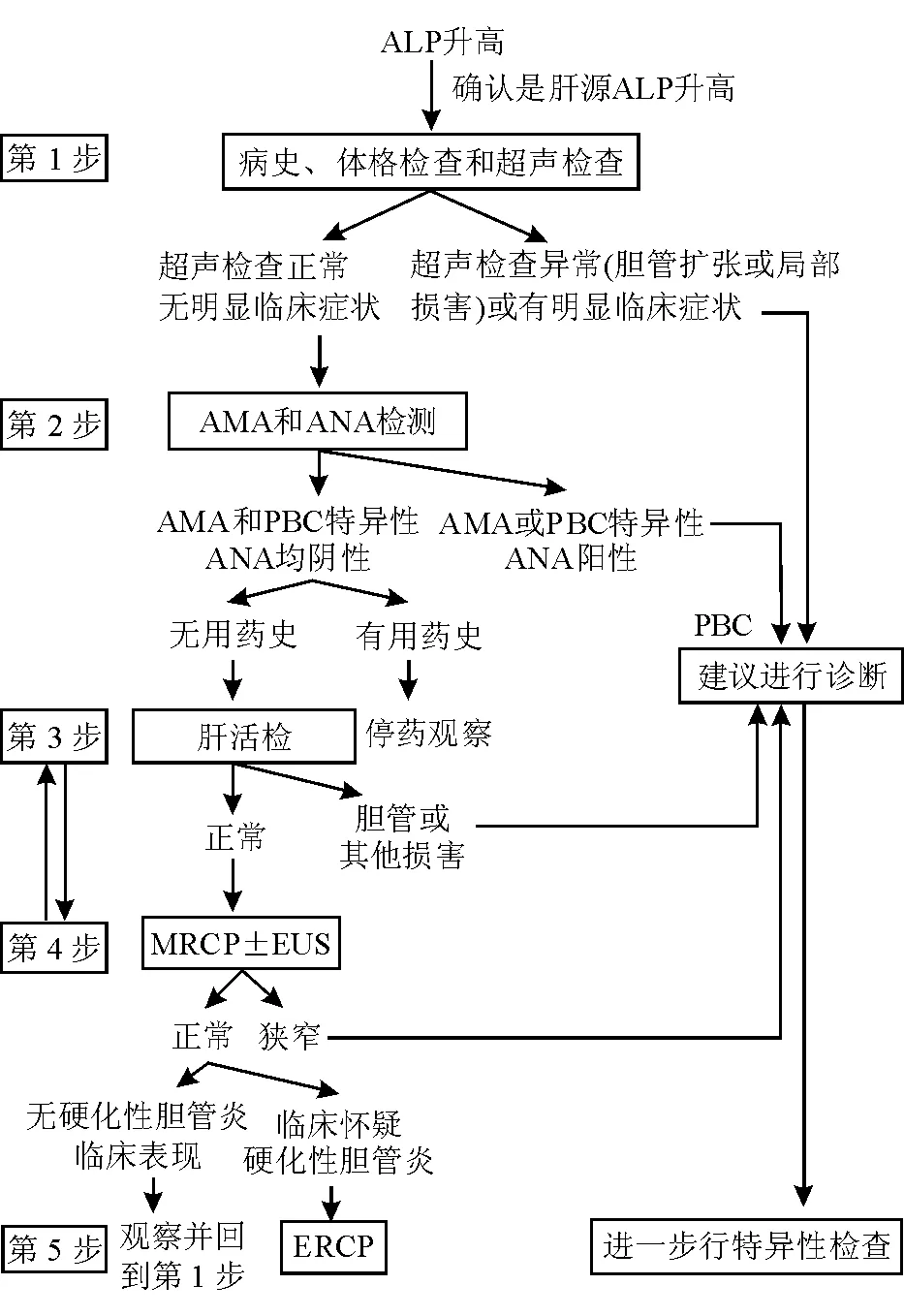
图1 胆汁淤积性肝病的诊断流程图
推荐意见
1. 血生化检查发现ALP超过1.5倍ULN且GGT超过3倍ULN可诊断胆汁淤积性肝病(B1)。但在PFIC 1型和2型以及BRIC等,GGT可不升高(B2)。
2. 腹部超声是区分肝内和肝外胆汁淤积的首选影像学检查方法,胆总管扩张且内径超过8 mm提示肝外梗阻(C1)。当腹部超声检查不能明确胆管扩张时,推荐行MRCP 和(或)EUS检查,并检测病毒性肝炎标记物和肝病相关自身抗体,如AMA等(C1)。
3. MRCP或EUS检查不能明确诊断时可考虑行ERCP(A1)。
4. 对无法解释的肝内胆汁淤积且AMA阴性者应考虑肝组织活检(C1)。对AMA阴性和肝组织活检未能确诊者,有条件者可考虑基因检测(C1)。
5. 胆汁淤积早期仅胆汁淤积相关酶学指标(ALP和GGT)和胆汁酸升高,可不出现黄疸。黄疸患者需排除遗传性高胆红素血症和血液系统疾病(B1)。
八、治疗
1. 治疗原则:去除病因和对症治疗。最有效的治疗是病因治疗,如手术或经内镜取结石,手术切除肿瘤,对PBC和PSC可用UDCA,对药物性和酒精性肝病(alcoholic liver disease, ALD)及时停用有关药物和戒酒最为重要,乙型和丙型病毒性肝炎予抗病毒治疗,AIH可用皮质激素取得缓解。
2. 药物治疗:药物治疗的目的是改善由胆汁淤积所致的临床症状和肝损伤。主要治疗药物为UDCA和S-腺苷蛋氨酸(S-adenosyl-L-methionine, SAM)。UDCA可用于治疗PBC、PSC、ICP、囊性肝纤维化、肝移植术后胆汁淤积、药物性胆汁淤积、PFIC和Alagille综合征等[2,13-14]。UDCA一般剂量为 10~15 mg·kg-1·d-1,Byler病和Alagille综合征可增至 45 mg·kg-1·d-1,囊性肝纤维化为 20~25 mg·kg-1·d-1。SAM可用于肝细胞性胆汁淤积、ICP和药物性胆汁淤积。初始治疗,肌内或静脉注射SAM,每日0.5~1.0 g,共2周。维持治疗,口服SAM片,每日1.0~2.0 g[15-16]。免疫介导的胆汁淤积可考虑应用肾上腺糖皮质激素,但需充分权衡治疗收益和可能的不良反应。其他正在研究且有应用前景的治疗药物包括奥贝胆酸和非诺贝特类药物[17-18]。
3. 其他治疗:重度黄疸或严重瘙痒经积极内科治疗无效者可考虑应用非生物型人工肝方法治疗,主要包括血浆置换、胆红素吸附、血浆滤过透析和分子吸附再循环系统等,这些治疗方法需有经验的专科医师指导,并需患者和家属知情同意;对于患者远期生存的影响尚需进一步研究。经药物和上述治疗无效且出现肝功能衰竭者可考虑肝移植。有胆管狭窄或梗阻者可行ERCP干预治疗,如球囊扩张、内镜下乳头括约肌切开以及支架置入。
推荐意见
6. UDCA和SAM可治疗多种肝病所致的胆汁淤积,UDCA治疗剂量为10~15 mg·kg-1·d-1(A1)。SAM初始疗法为每日0.5~1.0 g肌内或静脉注射,共2周;维持治疗为口服SAM片每日1.0~2.0 g(B1)。
7. 免疫介导的胆汁淤积需充分权衡治疗收益和可能的不良反应,可使用肾上腺糖皮质激素(B1)。积极内科治疗无效时可考虑应用非生物型人工肝方法治疗(C1),严重肝功能衰竭者可考虑肝移植(A1)。
九、常见胆汁淤积性肝病的诊断和治疗
PBC、PSC、AIH和药物性胆汁淤积见相关诊治共识或指南,不再赘述。
(一)病毒性肝炎合并胆汁淤积
1. 诊断:各型病毒性肝炎均可引起胆汁淤积,可分为急性、慢性两型。胆汁淤积型肝炎(或称淤胆型肝炎)是病毒性肝炎的一个临床类型,其临床表现与肝外梗阻性黄疸相似,有些病例很难与之鉴别。该型肝炎起病时自觉症状常较轻,以戊型肝炎病毒感染多见,进而可有皮肤瘙痒和疲劳,常有明显肝脏肿大,除ALT、AST、GGT和ALP水平升高外,还可有胆红素明显升高,以结合胆红素为主。慢性肝炎基础上发生上述临床表现者,可诊断为慢性淤胆型肝炎。急性淤胆型肝炎尚需与重型肝炎和妊娠急性脂肪肝相鉴别。妊娠急性脂肪肝发生于妊娠晚期,尿胆红素往往阴性,尿胆原阳性,超声检查示明显脂肪肝,易与淤胆型肝炎区别。病毒性肝炎合并胆汁淤积者,若见“酶疸分离”现象,临床上易误诊为重型肝炎,但两者的处理和转归各异。重型肝炎黄疸加深常与肝功能损害平行发展,消化道症状也较明显,尤其是凝血酶原活性呈进行性下降,可进行鉴别。诊断依据为病毒性肝炎血清标记物阳性,ALP和GGT升高,并排除其他原因所致者。
2. 治疗:强调病因治疗,在此基础上进行保肝、改善胆汁淤积治疗。慢性乙型和丙型病毒性肝炎的治疗关键是抗乙型肝炎病毒(HBV)和抗丙型肝炎病毒(HCV)治疗,只要有适应证且条件允许,就应进行规范的抗病毒治疗[19-20]。抗HBV治疗可予核苷(酸)类似物或干扰素,抗HCV治疗可予干扰素联合利巴韦林、直接抗病毒药物(DAAs)蛋白酶抑制剂、聚合酶抑制剂等药物。急性病毒性肝炎多能自愈,无须特殊药物治疗,患者需适当休息,平衡饮食。有些患者出现明显肝功能异常甚至黄疸,可给予适当的药物治疗。胆汁淤积明显者可选用UDCA和(或)SAM。
推荐意见
8. 各型肝炎病毒感染均可引起胆汁淤积型肝炎,急性病毒性肝炎多能自愈,无须特殊药物治疗。慢性乙型和丙型肝炎治疗上首选抗HBV和抗HCV的药物,有胆汁淤积者可选用UDCA和(或)SAM(B1)。
(二)ALD合并胆汁淤积
1. 诊断:ALD是由长期大量饮酒导致的中毒性肝损伤,初期为酒精性脂肪肝,进而发展为酒精性肝炎、肝纤维化,最终导致肝硬化。各型ALD均可发生胆汁淤积。结合饮酒史(一般饮酒时间>5年,乙醇摄入量男性≥40 g/d,女性≥20 g/d,或2周内有大量饮酒史,乙醇摄入量>80 g/d)、临床表现、实验室和影像学检查常可明确ALD合并胆汁淤积的诊断[21]。肝组织活检仅用于少数疑难病例,旨在探明病因、病变程度、判断预后和指导治疗。如怀疑肝外胆管梗阻,则需进一步行MRCP、ERCP 和(或)PTC 等检查。
2. 治疗:治疗原则是戒酒和营养支持治疗,减轻ALD的严重程度,改善已存在的继发性营养不良和对症治疗酒精性肝硬化及其并发症。应用最广泛的ALD治疗药物为激素,但需排除胰腺炎、消化道出血、肾功能衰竭或活动性感染。对合并胆汁淤积的重症病例,推荐使用肾上腺皮质激素治疗,如使用激素7 d黄疸无消退,提示无应答,应停用激素,避免不良反应的发生。美国肝病研究学会(AASLD)的《酒精性肝病诊疗指南》[22]推荐的预后评估方法是Maddrey判别函数(Maddrey discriminant function, MDF),MDF=4.6×(患者凝血酶原时间-对照凝血酶原时间)+总胆红素(mg/dL)。激素使用的阈值(MDF评分≥32 定义为高风险病死率)可能存在一个最大限度,超过这个阈值,减少炎症级联反应的内科治疗可能弊大于利。当MDF评分>54时,使用激素者较未使用者有更高的病死率。AASLD指南建议激素治疗的剂量和疗程分别为40 mg/d和4周,然后减量维持2~4周或停药观察,同时给予UDCA和(或)SAM治疗。
推荐意见
9. ALD合并胆汁淤积提示预后不良,戒酒是ALD最主要的治疗措施,同时应重视营养支持治疗(B1)。合并胆汁淤积的重症病例,如MDF评分≥32,且排除胃肠道出血、细菌感染等激素禁忌证,推荐使用肾上腺皮质激素40 mg/d治疗4 周,然后减量维持2~4周或停药,并应用UDCA和(或)SAM(B1)。
(三)NAFLD合并胆汁淤积
1. 诊断:NAFLD可有临床生化上的胆汁淤积表现以及组织学肝内胆管病变,但这并不是NAFLD的特征性表现,而更常见于一些继发于特殊病因的NAFLD,如全胃肠外营养、药物/毒物、丙型肝炎、空-回肠旁路减肥术后和肝豆状核病变等[23]。胆汁淤积本身与这些致病因素相关。NAFLD发展到肝硬化、肝功能衰竭等终末期肝病阶段,或合并其他类型的肝病时也可有胆汁淤积表现。部分NAFLD在单纯性脂肪肝和NASH阶段也有血清GGT、ALP和TBA轻中度升高。伴有ALP和GGT显著升高的NAFLD患者有肝组织学肝细胞或胆管病变,NAFLD出现胆汁淤积可能提示肝损伤更严重。
2. 治疗:去除病因和诱因,治疗原发基础疾病或伴随疾病,改变生活方式,如节食、运动、禁酒、戒烟;发展至失代偿期肝硬化和肝功能衰竭及其并发症时肝移植可能是惟一有效的治疗选择。伴有胆汁淤积者选用UDCA和(或)SAM。
推荐意见
10. 首先去除病因和诱因,并以饮食、运动和行为纠正非药物治疗为主(C1)。同时根据患者代谢紊乱特点进行个体化治疗(如改善胰岛素抵抗、降血脂和减重等),可选用UDCA和(或)SAM(C1)。
(四)遗传性胆汁淤积性肝病
1. 囊性纤维化相关的肝病(cystic fibrosis-associated liver disease, CFALD):CFALD是一种常染色体隐性遗传性疾病,由第7染色体长臂上的跨膜转导调节蛋白(CFTR)基因突变引起。表现为肝肿大、至少两项肝功能检查持续升高和超声检查异常,可伴有先天性胆汁淤积、肝脂肪变、局灶或多叶性肝硬化。CFALD 并发症是囊性纤维化相关死亡第二常见的原因[24]。①诊断:CFALD 尚未有明确的诊断标准。1/3患者有肝肿大,其可由CFALD或肺心病肝充血所致。ALP、ALT、AST、胆红素和GGT持续异常超过1.5倍ULN时应进一步检查(如凝血酶原时间、白蛋白)以严格评估肝损伤并排除其他原因的肝病(如药物、毒素、感染、胆管闭锁、胆石症、抗胰蛋白酶缺乏症、AIH、PSC或其他原因的胆管梗阻)。超声检查可见CFALD的征象,如肝肿大或胆管异常,肝脏内见数量不等、大小不一的无回声占位病变。CT检查可见囊肿大小和残存正常肝组织。由于许多患者伴有局灶性纤维化、肝硬化,肝组织活检意义不大[25]。②治疗:尚未发现对CFALD预后有益的治疗药物。推荐优化胆汁淤积患者的营养状态,以避免维生素缺乏和营养不良,但未被证明有效。予20~30 mg·kg-1·d-1UDCA可持续改善肝功能、刺激受损胆管分泌胆汁、改善组织学指标(2年以上)和营养状态[26-27]。该病患者一般寿命长,预后常取决于同时存在的肾囊肿的严重程度,极少癌变。本病很少需行手术治疗,有急性症状时可在超声指导下穿刺抽液,但囊液可重新产生。日常生活严重受限或终末期患者可考虑肝移植[28]。
推荐意见
11. 长期随访发现,CFALD可发生于1/3的囊性纤维化患者。肝肿大、肝功能异常和肝脏超声检查示数量不等、大小不一的无回声占位病变可作出诊断(C2)。未证实对CFALD有长期疗效的治疗方法(C2)。UDCA(20~30 mg·kg-1·d-1)可改善CFALD患者的肝功能和组织学指标(C1)。日常生活严重受限或终末期患者应考虑肝移植治疗(B1)。
2. PFIC:PFIC是一种以慢性胆汁淤积为特征的常染色体隐性遗传性疾病,由毛细胆管转运蛋白基因ATP结合盒(ATP-binding cassette, ABC)转运蛋白基因突变所致[29]。婴儿期发病,通常在10年内进展至肝硬化[30]。①分型:PFIC分为3种类型, PFIC 1型,既往称作Byler病,主要由ATP8B1基因突变所致。肝病的典型症状和体征见于新生儿期。相较于血清转氨酶、胆红素和胆汁酸升高而言,血清GGT较低(相较于胆管闭锁和Alagille综合征)。肝组织学检查提示纤维化,但无胆管增生,大多数患者在12岁前进展至终末期肝病。腹泻、胰腺炎、发育障碍和听力缺失是ATP8B1基因突变的肝外表现。PFIC 2型既往称作“Byler综合征”,主要由ABCB11基因突变所致,ABCB11基因编码胆汁酸盐输出泵蛋白(BSEP)。幼儿期临床表现、生化检查和进展期肝病的症状类似于PFIC 1型,GGT水平较低。肝组织学检查提示门静脉炎症和巨细胞肝炎。电镜检查显示,PFIC 1型有粗颗粒胆汁而PFIC 2型有液状胆汁。PFIC 3型由编码毛细胆管磷脂转运体ABCB4-MDR3的ABCB4基因突变所引起,与1 型和2型不同,PFIC 3型患者GGT通常明显升高,肝组织学检查除发现门静脉炎症和纤维化/肝硬化外,尚有弥漫性胆管增生。PFIC 3型可伴发肝内胆石症。②治疗:未发现有对PFIC长期预后有益的治疗方法。对儿童患者,普遍推荐予补充中链三酰甘油和脂溶性维生素。UDCA可改善50% PFIC 3型患者的生化指标,但对1型和2型无效[30]。利福平可缓解瘙痒。据报道,部分胆管分流和回肠旷置术可改善PFIC 1型和2型的症状和体征[31]。对晚期PFIC,建议肝移植治疗。
推荐意见
12. PFIC 1、2 和3型均为少见的发生于幼儿和青少年期的慢性进展性胆汁淤积性肝病。PFIC 1 型和2型以低GGT水平、严重瘙痒和各种肝外表现为特征。PFIC尚无有效的治疗方法(C2)。UDCA可改善部分PFIC 3型患者血清肝功能指标(C2)。部分胆汁分流对PFIC 1型和2型的临床表现和生化指标有益(C2)。晚期患者推荐行肝移植治疗(B1)。
3. BRIC:以反复发作的自限性严重瘙痒、胆汁淤积和黄疸为特征,可持续数周至数月,常有数月或数年的无症状期。每次发作不会发生进行性肝损伤和肝硬化。肝组织学检查显示,胆汁淤积伴胆管阻塞、门管区扩张、单核细胞浸润和某些肝细胞变性。缓解期肝组织学和肝功能正常。BRIC 1型和2型为青少年和成人急性胆汁淤积性疾病,是PFIC 1型和2型的良性表现形式,主要由ATP8B1和ABCB11基因错义突变引起[32]。BRIC 1 型可伴有胰腺炎,而BRIC 2型可伴发胆石症。BRIC的诊断标准[33]:①持续数月至数年的无症状间隔黄疸至少发作2次;②实验室检查符合肝内胆汁淤积;③GGT正常或仅轻微升高;④继发于胆汁淤积后的严重瘙痒;⑤肝组织学检查证实小叶中心性胆汁淤积;⑥胆管造影术显示肝内或肝外胆管正常;⑦没有已知的其他导致胆汁淤积的因素(如药物和妊娠等)。诊断关键是至少6个月的无症状、间隔性、多次黄疸发作,且无药物或毒性物质接触史或胆管疾病等诱因。因此BRIC与其他原因导致的慢性胆汁淤积和瘙痒的鉴别诊断相对直接明了。BRIC病因不明,没有预防和限制发作病程的特异性治疗。治疗的关键是缓解症状直至瘙痒和其他症状自然消退。胆汁淤积引发严重瘙痒的替代治疗是利福平,次选苯巴比妥。予BRIC患者UDCA治疗后增加了胆汁酸池,同时增加了UDCA在胆汁酸池中的比例,降低毒性疏水性胆汁酸在胆汁中的比例。治疗PBC时,UDCA标准剂量为13~15 mg·kg-1·d-1,治疗其他胆汁淤积肝病时,剂量可增至2倍。治疗BRIC的适合剂量尚不明。有些报道提示UDCA可改善BRIC患者的瘙痒症状,缩短发作病程并预防再次发作。考虑到BRIC患者发作病程的变化和无症状间隔期,很难确定是UDCA还是其他治疗方法有效。SAM已被用于治疗伴有妊娠、服用雌激素以及其他药物所致的胆汁淤积,其作用机制尚不清楚。但在一项单盲交叉研究[34]中,BRIC患者静脉注射SAM并未改善瘙痒症状,也未能改善生化指标异常。有报道显示鼻胆管引流可缓解BRIC[35]。反复发作的BRIC有发展为PFIC的可能。
推荐意见
13. BRIC以反复发作的自限性严重瘙痒、胆汁淤积和黄疸为特征,可持续数周至数月,常有数月或数年无症状期(C1)。尚无BRIC的有效治疗方法,UDCA或鼻胆管引流治疗处于试验阶段(C2)。
4. Alagille综合征:主要发生于儿童和青少年,表现为轻重不一的胆汁淤积。临床表现以胆汁淤积伴瘙痒和小叶间胆管减少,心血管系统、眼、骨骼、面部异常为特征的多系统损害,其他少见的临床表现有肾功能损害、神经血管和胰腺异常等。病因是JAG1或Notch2基因缺失,包括整基因缺失、蛋白截短、剪接和错义突变。该基因编码对细胞分化起重要作用的Notch信号通路的配体。病变缺陷可能位于第20号染色体短臂[36]。诊断主要依据Alagille提出的标准[37]:①肝组织学检查见小叶间胆管减少;②五大临床特征(包括慢性胆汁淤积、心脏疾病、骨骼异常、眼睛异常和特征性面部异常)中至少存在三项;其他系统表现少见,主要侵犯肾脏和神经血管系统。目前尚无有效的治疗方法。有报道部分胆管分流可缓解严重的瘙痒症状[2]。
推荐意见
14. Alagille综合征主要发生于儿童和青少年,由JAG1或Notch2基因缺失导致胆汁淤积伴瘙痒和小叶间胆管减少,心血管系统、眼、骨骼、面部异常为特征的多系统损害,尚无有效的治疗方法,部分胆管分流可缓解严重的瘙痒症状(C2)。
(五)ICP
ICP 的发病机制涉及多种因素,遗传、激素和环境因素起重要作用。患ICP后,经母体流向胎儿的胆汁酸增加,表现为胆汁酸在羊水、脐带血和胎粪中升高。孪生妊娠时ICP发病率增高,大剂量服用避孕药和孕酮可诱发ICP,支持激素在该病发生中起关键作用的观点[38]。同一家族成员ICP发病率增加以及种族间差异说明了遗传因素的作用。最近遗传学研究鉴定出毛细胆管转运蛋白基因变异体(ABC转运体B4=磷脂酰胆碱转移酶,ABC转运体B11=胆酸盐输出泵,ABC转运体C2=结合型有机离子转运体,ATP8B1=FIC1)和某些ICP患者调节物[胆酸盐感受器法尼醇X受体(FXR)]。妊娠过程中,当激素和其他底物超过毛细胆管转运体转运能力时,毛细胆管转运体轻度功能不全就能诱发胆汁淤积。目前,遗传学检测仅限于实验研究,并不用于诊断或危险性分级。因此如产后胆汁淤积(伴有GGT 水平升高)持续存在,可考虑行ABCB4基因突变分析。
1. 诊断:ICP是病因不明的妊娠特发性、可逆的胆汁淤积性疾病,出现于妊娠晚期,分娩后自行迅速改善。其特征为:①妊娠期有严重瘙痒(多起始于妊娠第二或第三阶段);②血清ALT、空腹血清胆汁酸和甘胆酸升高;③产后(4~6周内)症状和体征自行缓解[38]。瘙痒导致孕妇不适和烦恼,ICP也增加早产和胎儿猝死风险。ICP一般预后较好,但黄疸久者,因缺乏维生素K依赖性凝血因子Ⅱ、Ⅶ、Ⅹ,生产时可发生大出血。本病对胎儿影响较大,发生胎儿宫内窘迫、早产和死胎的危险性相对较高。胆汁酸是妊娠肝内胆汁淤积最敏感的指标,可先于其他指标出现异常。确诊ICP可根据临床表现结合血清甘胆酸和TBA水平,血清甘胆酸≥10.75 μmol/L或TBA≥10 μmol/L可诊断ICP[39]。妊娠期间胆汁酸水平≥40 μmol/L与胎儿并发症增加相关。ABCB4基因变异的ICP患者GGT水平增高,否则GGT正常。10%~15%患者血清结合胆红素中度升高,出现轻度黄疸。ICP诊断要求排除其他疾病,如皮肤疾病、肝胆系统疾病(肝炎病毒、EB病毒、巨细胞病毒和ABCB4基因缺陷症等导致的肝损害以及自身免疫性肝病),且通过产后随访作出最后诊断。超声检查有助于排除其他严重肝病和肝胆系统结石,肝组织活检通常是非必需的。妊娠期血清肝功能检查异常除ICP外,尚需考虑先兆子痫(HELLP综合征)和妊娠急性脂肪肝[39]。HELLP综合征以溶血、肝酶升高和血小板减少(常<50×109/L)为特点,是妊娠期高血压的严重并发症,多发生在产前,胆红素多<5 mg/dL(<85 μmol/L),影像学显示肝坏死、血管瘤、肝破裂。妊娠急性脂肪肝是一种妊娠晚期的急性肝脂肪变,多累及年轻初产妇,在妊娠最后三个月或产后早期发生,起病急骤,预后凶险,临床表现如同急性重型肝炎,表现为急性肝功能衰竭,常伴有肾功能衰竭;影像学检查显示肝脏脂肪变。产后出现持续性异常应考虑其他慢性肝病,如PBC、PSC、ABCB4基因缺失或慢性丙型肝炎,这些疾病可能在妊娠晚期出现瘙痒。
2. 治疗:随机临床研究表明,UDCA(10~20 mg·kg-1·d-1)可作为治疗ICP的一线药物,可改善67%~80% ICP患者的瘙痒和血清肝功能指标[40-41]。但有研究显示,由于胎儿并发症发病率低,分别应用UDCA和安慰剂治疗的患者的并发症降低率并无差异。SAM疗效逊于UDCA,但可能有附加效果[15-16]。如经数天UDCA标准治疗后瘙痒无减轻,可选择SAM或根据个体情况考虑使用利福平。局部搽剂是安全的,但疗效不清楚。药物治疗的同时,还需注意产科情况。强调治疗过程中加强胎儿监护,把握终止妊娠的时机,对降低围生儿死亡率具有重要意义。妊娠35周后,若出现病情进展、宫缩不能抑制、胎动异常、胎心率变异或应激试验无反应、羊水胎粪污染等,应把握时机,积极终止妊娠[42-43]。
推荐意见
15. ICP诊断依据:①妊娠期瘙痒;②血清ALT 水平以及空腹TBA和甘胆酸水平升高;③除外其他原因的肝功能异常或瘙痒。产后肝功能完全正常后,可确诊ICP(B2)。
16. ICP 产妇自发性或医源性早产率增加(B1)。UDCA和SAM可用于妊娠第二或第三期胆汁淤积且有症状的患者,缓解瘙痒并能改善血清肝功能指标(B1),但缺乏有关胎儿保护和并发症减少的数据(C2)。凝血酶原时间延长时应补充维生素K(C2)。分娩时间应根据个体情况决定(C2)。
十、肝外表现和处理
(一)瘙痒
瘙痒是胆汁淤积的并发症之一,是一种仅有皮肤不适感觉而无原发性皮肤损害的症状,这种感觉无论在性质、持续性和定位上均不同于触觉和痛觉。胆汁淤积性瘙痒的发病机制目前尚不清楚[44],大多数学者认为瘙痒可能与血清自分泌运动因子(autotaxin)活性增加和溶血磷脂酸形成有关。此外,胆汁酸盐、内源性阿片肽、5-羟色胺(5-HT)、感觉神经元过度兴奋、雌激素和孕激素、肝肠瘙痒原改变、遗传因素(如ICP)等也可能与瘙痒有关[45]。瘙痒的严重度评分有下列三种:①视觉模拟评分(visual analogue scale, VAS):瘙痒的严重度按皮肤抓痕分为:抓痕、斑块、结节和(或)瘢痕,根据轻、中、重度分别评分0~3(4)分,总分从0分无瘙痒到10分严重瘙痒[46];②瘙痒严重程度量表(itch severity scale, ISS):包括频率、睡眠、心情、性欲、性功能、李克特量表(Likert scale)评估瘙痒强度、瘙痒涉及的体表面积7项,总评分范围可从0分无瘙痒到21分最严重的瘙痒[47];③半定量评估瘙痒:根据瘙痒频率分为4个阶段:偶尔瘙痒、无临床症状的每天间断性瘙痒、出现临床症状的每天间断性瘙痒和持续性瘙痒[48]。
多种药物可单独或联合应用治疗胆汁淤积性肝病瘙痒,包括考来烯胺(消胆胺)、抗组胺药、孕烷X受体激动剂、阿片受体拮抗剂、5-HT受体拮抗剂,但具体的治疗机制仍未明确。考来烯胺是一种具有降低血清胆固醇水平作用的不吸收阴离子交换树脂,可减低胆汁酸肠肝循环,从而降低血清胆汁酸水平,减轻瘙痒。治疗瘙痒推荐剂量为 4 g/d,不超过16 g/d。考来烯胺有异味,并可引起消化道症状,如便秘、腹胀等,因而耐受性较差,长期用药可加重脂肪泻,致脂溶性维生素缺乏。UDCA和SAM在治疗胆汁淤积过程中对瘙痒症状也有改善作用。一项纳入84例ICP瘙痒患者的随机研究[48]显示,分别使用UDCA、考来烯胺治疗瘙痒14 d,UDCA组症状改善有效率达66.6%,显著高于考来烯胺组(19.0%,P<0.05),且UDCA组瘙痒评分的改善显著优于考来烯胺组。对46例ICP患者分别给予UDCA、SAM口服治疗的研究[16]显示,瘙痒评分均有不同程度的改善。孕烷X受体激动剂利福平也可改善瘙痒,可作为治疗药物[49-50]。利福平一般以150 mg/d 单剂口服,有效后继续服用。如无效,可隔周递增至600 mg/d。利福平可改善继发于PBC的胆汁淤积性瘙痒,其确切机制尚未阐明。但有研究显示,用利福平治疗美沙酮(一种镇痛药)成瘾患者时可引起阿片戒断反应,因此利福平可能存在阿片拮抗作用,进而缓解胆汁淤积性瘙痒。值得注意的是,使用利福平后患者尿色变红,还可出现中毒性肾损害,偶有溶血发生。同时,治疗瘙痒剂量的利福平可有肝脏毒性,因此用药期间必须随访肝功能。此外,利福平可通过干扰维生素D的吸收而加重骨质软化;通过超敏反应引发胆汁淤积性肝炎,甚至发生胆汁淤积性瘙痒等并发症。事实上,由于利福平潜在的不良反应,胆汁淤积性肝病瘙痒的治疗应慎用。口服阿片受体拮抗剂纳曲酮25 mg/d 可用于治疗瘙痒,少数患者可有恶心、呕吐、轻度疼痛等不良反应。纳曲酮有一定程度的肝脏毒性,使用时需监测肝功能。纳曲酮的代谢产物可在失代偿期肝病患者体内积聚,因此这些患者使用时需谨慎[50]。这类药物应先小剂量使用,再逐渐提高剂量,以免引起类似麻醉药的戒断反应。如上述药物均无效可应用选择性5-HT再摄取抑制剂舍曲林治疗,初始剂量建议50 mg/d,数周后可增至100 mg/d[51]。近年来,紫外线照射、血浆置换、体外白蛋白透析和鼻胆管引流也用于改善胆汁淤积性瘙痒,并获得较好的疗效。肝移植可用于药物和其他方法疗效不佳的患者。
推荐意见
17. 考来烯胺治疗瘙痒的推荐剂量是4 g/d,不超过16 g/d,与UDCA和其他药物服用的间隔至少4 h(B1)。
18. 阿片受体拮抗剂纳曲酮治疗瘙痒先小剂量口服25 mg/d,无效再逐渐提高至 50 mg/d,以免引起类似麻醉药的戒断反应(C1)。舍曲林治疗瘙痒初始剂量为50 mg/d,数周后可增至100 mg/d(C2)。
19. 利福平治疗瘙痒一般以150 mg/d 单剂口服,有效后继续服用。如无效,可隔周递增至600 mg/d,用药期间须随访肝功能。由于利福平潜在的不良反应,对于其在胆汁淤积性肝病患者瘙痒的治疗中应慎用(B2)。
20. 上述治疗无效者可考虑选用紫外线照射、血浆置换、体外白蛋白透析和鼻胆管引流等,可改善胆汁淤积性瘙痒(C2)。药物和其他方法疗效不佳的患者可考虑肝移植(C2)。
(二)疲劳
胆汁淤积患者常有疲劳症状,尤其是PBC。疲劳是一个复杂的症状,包括持续的衰竭感觉、正常工作能力缺失、心理和生理功能下降。由于非特异性且缺乏客观的评估方法,因此疲劳至今仍被忽视。目前对疲劳的发病机制及其治疗仍不清楚[52],尚无有效改善胆汁淤积性肝病患者疲劳症状的治疗方法。需注意的是,治疗前需排除贫血、糖尿病、甲状腺功能减退、肾和肾上腺功能不全、抑郁等。目前可能的治疗药物有选择性5-HT3受体拮抗剂,如昂丹司琼、阿片受体拮抗剂和中枢神经兴奋药莫达非尼等治疗,莫达非尼起始剂量为 100 mg/d,根据患者耐受情况和对药物的反应剂量逐渐增至 200 mg/d,但其疗效有待进一步研究证实[53-54]。UDCA是治疗PBC的有效药物,可延缓疾病进程,延长移植前的生存时间,然而对伴随的疲劳症状无明显改善作用。肝移植不能降低疲劳的发生,甚至在移植1 年后,疲劳仍是使患者痛苦的症状,但程度减轻。避免刺激,提倡健康的生活方式,包括足够的睡眠、规律锻炼、戒酒和戒咖啡等均有好处。抗抑郁药可部分减轻抑郁患者的疲劳。
推荐意见
21. 排除贫血、糖尿病、甲状腺功能减退、肾和肾上腺功能不全、抑郁等,足够的睡眠、规律锻炼、戒酒和晚上戒咖啡等对患者改善疲劳症状有益(C2)。
22. 昂丹司琼、阿片受体拮抗剂和莫达非尼(100~200 mg/d)等的疗效有待证实,抗抑郁药可部分减轻抑郁患者的疲劳(C2)。肝移植对疲劳改善无明显作用(C1)。
(三)骨质疏松
骨质疏松以单位体积骨组织总量减少为特征,骨松质表现为骨小梁明显减少、变薄,骨密质表现为骨皮质变薄、疏松。骨质疏松可引起骨折,尤其易发生在椎体、髋骨和前臂。终末期肝病和重度胆汁淤积患者骨质疏松风险增加,发生率为9%~60%,可能与维生素D缺乏、营养不良等有关。骨质疏松治疗包括健康的生活方式、补充钙剂和维生素D以及药物干预。应坚持健康的生活方式,包括摄入富含维生素D、钙、低盐和适量蛋白质的均衡膳食,避免嗜烟、酗酒,慎用影响骨代谢的药物,进行适度的肌力锻炼和康复治疗。国外推荐剂量为元素钙1 500 mg/d,维生素D 800 IU/d。我国营养协会推荐成人每日钙摄入量800 mg(元素钙);绝经后妇女和老年人每日钙摄入推荐量为1 000 mg。维生素D的成年人推荐剂量为200 IU/d;老年人因缺乏日照以及摄入和吸收障碍,故推荐剂量为 400~800 IU/d。维生素D 用于治疗骨质疏松时,剂量应为800~1 200 IU/d。流行病学资料支持补钙(1 000~1 200 mg/d)和增加维生素D(400~800 IU/d)的摄入,可减少或逆转骨质自然丢失的速度,但无支持或反对的实验证据。激素替代疗法对绝经后的女性有效[55]。由于存在诱发肝细胞癌的风险,男性患者应避免使用睾酮。研究证据支持使用二膦酸盐类如阿仑膦酸钠(10 mg/d)治疗骨质疏松[56],支持雷洛昔芬和氟化钠的数据有限[57]。骨矿密度测量可用于骨质疏松的治疗和随访[58]。
推荐意见
23. 建议患者补充钙和维生素D预防骨质疏松。成人每日钙摄入量800 mg ;绝经后妇女和老年人每日钙摄入推荐量为1 000 mg。维生素D的成年人推荐剂量为200 IU/d ;老年人推荐剂量为400~800 IU/d(C1)。支持阿仑膦酸钠(10 mg/d)治疗骨质疏松,支持雷洛昔芬和氟化钠的数据有限(C2)。每年骨矿密度测量可用于骨质疏松的治疗和随访(C2)。
(四)脂溶性维生素缺乏
胆汁淤积时肝脏分泌胆汁至小肠发生障碍,肠内胆盐减少,可出现脂溶性维生素缺乏和脂肪泻,因此需适当补充脂溶性维生素[2]。如凝血酶原时间延长,肌内注射维生素K1(10 mg/d)直至正常。因维生素A缺乏所致的夜盲,可口服维生素A 25 000~50 000 IU/d。维生素E缺乏少见,可口服予以补充。建议测定血液脂溶性维生素水平以指导其补充,但目前尚未得到普遍使用和推广。
推荐意见
24. 凝血酶原时间延长,肌内注射维生素K1(10 mg/d)直至正常。维生素A缺乏所致的夜盲,可口服维生素A 25 000~50 000 IU/d。维生素E缺乏少见,可口服10~100 mg/d予以补充。维生素D补充 400~800 IU/d(C2)。
十一、待解决的问题
尽管近年来在胆汁淤积性肝病的诊断和治疗方面有不少进展,但该领域仍面临诸多问题和挑战。有关胆汁淤积的发生机制,尤其是分子机制尚未阐明;胆汁酸转运蛋白遗传和变异对胆汁淤积性肝病发生和发展的影响、胆汁淤积中胆汁酸的成分及其对肝脏和全身的影响有待进一步研究;胆汁淤积性肝病诊断标记物和诊断标准需进一步完善和验证;治疗上尚需寻找更有效的药物和方法等。
参加本共识撰写和讨论的专家名单(排名不分先后,按姓氏汉语拼音为序):
陈成伟成军窦晓光段钟平范建高
傅青春高春芳韩涛韩英侯金林
胡和平胡锡琪黄建荣贾继东刘玉兰
陆伦根马雄茅益民南月敏牛俊奇
邱德凯任红尚佳唐红王贵强
王吉耀王建设王磊王宇明魏来
谢青谢渭芬许建明徐铭益徐小元
杨长青杨云生尤红曾民德张文宏
张跃新周新民庄辉邹晓平
参考文献
1 Kuntz K, Kuntz HD. Cholestasis[A]// Hepatology: principles and practice[M]. 2ndedition. Springer Medizin Verlag Heidelberg, 2006: 227-242.
2 European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases[J]. J Hepatol, 2009, 51 (2): 237-267.
3 Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations[J]. BMJ, 2008, 336 (7650): 924-926.
4 Bortolini M, Almasio P, Bray G, et al. Multicentre survey of the prevalence of intrahepatic cholestasis in 2520 consecutive patients with newly diagnosed chronic liver disease[J]. Drug Investigation, 1992, 4 (Suppl 4): 83-89.
5 Cheng J, Zhang WH, Wang JB, et al. A cross-sectional study on intrahepatic cholestasis indicators of viral hepatitis patients[A]. 2015, EASL. P0592.
6 曹旬旬, 高月求, 张文宏, 等. 基于上海市住院慢性肝病患者胆汁淤积患病率的调查研究[J]. 中华肝脏病杂志, 2015, 23 (8): 569-573.
7 Burt A, Portmann B, Ferrell L. MacSween’s pathology of the liver[M]. 6thedition. Elsevier Churchill Livingstone, 2012: 503-562.
8 Bacon BR, O’Grady JG, Di Bisceglie AM, et al. Comprehensive clinical hepatology[M]. 2ndedition. Elsevier, 2006: 87.
9 Siddique A, Kowdley KV. Approach to a patient with elevated serum alkaline phosphatase[J]. Clin Liver Dis, 2012, 16 (2): 199-229.
10Geller S, Petovic LM. Evaluation of cholestasis//Biopsy interpretation of the liver[M]. 2ndedition. Lippincott Williams & Wilkins, 2009: 404-416.
11Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy[J]. N Engl J Med, 1996, 335 (13): 909-918.
12Heathcote EJ. Diagnosis and management of cholestatic liver disease[J]. Clin Gastroenterol Hepatol, 2007, 5 (7): 776-782.
13Beuers U. Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis[J]. Nat Clin Pract Gastroenterol Hepatol, 2006, 3 (6): 318-328.
14Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited[J]. Hepatology, 2002, 36 (3): 525-531.
15Binder T, Salaj P, Zima T, et al. Randomized prospective comparative study of ursodeoxycholic acid and S-adenosyl-L-methionine in the treatment of intrahepatic cholestasis of pregnancy[J]. J Perinat Med, 2006, 34 (5): 383-391.
16Roncaglia N, Locatelli A, Arreghini A, et al. A randomised controlled trial of ursodeoxycholic acid and S-adenosyl-l-methionine in the treatment of gestational cholestasis[J]. BJOG, 2004, 111 (1): 17-21.
17Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid[J]. Gastroenterology, 2015, 148 (4): 751-761.e8.
18Beuers U, Trauner M, Jansen P, et al. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond[J]. J Hepatol, 2015, 62 (1 Suppl): S25-S37.
19中华医学会肝病学分会, 中华医学会感染病学分会. 慢性乙型肝炎防治指南(2010年版)[J]. 中华肝脏病杂志, 2011, 19 (1): 13-24.
20European Association for Study of Liver. EASL clinical practice guidelines: management of hepatitis C virus infection[J]. J Hepatol, 2014, 60 (2): 392-420.
21中华医学会肝病学分会脂肪肝和酒精性肝病学组. 酒精性肝病诊疗指南(2010年修订版)[J]. 中华肝脏病杂志, 2010, 18 (3): 167-170.
22O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease[J]. Hepatology, 2010, 51 (1): 307-328.
23中华医学会肝病学分会脂肪肝和酒精性肝病学组. 非酒精性脂肪性肝病诊疗指南(2010年修订版)[J]. 中华肝脏病杂志, 2010, 18 (3): 163-166.
24Colombo C, Battezzati PM, Crosignani A, et al. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome[J]. Hepatology, 2002, 36 (6): 1374-1382.
25Sokol RJ, Durie PR. Recommendations for management of liver and biliary tract disease in cystic fibrosis. Cystic Fibrosis Foundation Hepatobiliary Disease Consensus Group[J]. J Pediatr Gastroenterol Nutr, 1999, 28 Suppl 1: S1-S13.
26Colombo C, Battezzati PM, Podda M, et al. Ursodeoxycholic acid for liver disease associated with cystic fibrosis: a double-blind multicenter trial. The Italian Group for the Study of Ursodeoxycholic Acid in Cystic Fibrosis[J]. Hepatology, 1996, 23 (6): 1484-1490.
27van de Meeberg PC, Houwen RH, Sinaasappel M, et al. Low-dose versus high-dose ursodeoxycholic acid in cystic fibrosis-related cholestatic liver disease. Results of a randomized study with 1-year follow-up[J]. Scand J Gastroenterol, 1997, 32 (4): 369-373.
28Swenson K, Seu P, Kinkhabwala M, et al. Liver transplantation for adult polycystic liver disease[J]. Hepatology, 1998, 28 (2): 412-415.
29Jacquemin E. Progressive familial intrahepatic cholestasis[J]. Clin Res Hepatol Gastroenterol, 2012, 36 Suppl 1: S26-S35.
30Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases[J]. Semin Liver Dis, 2001, 21 (4): 551-562.
31Arnell H, Bergdahl S, Papadogiannakis N, et al. Preoperative observations and short-term outcome after partial external biliary diversion in 13 patients with progressive familial intrahepatic cholestasis[J]. J Pediatr Surg, 2008, 43 (7): 1312-1320.
32Oude Elferink RP, Paulusma CC, Groen AK. Hepatocana-licular transport defects: pathophysiologic mechanisms of rare diseases[J]. Gastroenterology, 2006, 130 (3): 908-925.
33Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis[J]. Clin Liver Dis, 2004, 8 (1): 133-149, vii.
34Everson GT, Ahnen D, Harper PC, et al. Benign recurrent intrahepatic cholestasis: treatment with S-adenosyl-methionine[J]. Gastroenterology, 1989, 96 (5 Pt 1): 1354-1357.
35Stapelbroek JM, van Erpecum KJ, Klomp LW, et al. Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasis[J]. Hepatology, 2006, 43 (1): 51-53.
36Piccoli DA, Spinner NB. Alagille syndrome and the Jagged1 gene[J]. Semin Liver Dis, 2001, 21 (4): 525-534.
37Alagille D. Alagille syndrome today[J]. Clin Invest Med, 1996, 19 (5): 325-330.
38Lammert F, Marschall HU, Glantz A, et al. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management[J]. J Hepatol, 2000, 33 (6): 1012-1021.
39Hay JE. Liver disease in pregnancy[J]. Hepatology, 2008, 47 (3): 1067-1076.
40Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of cholestasis of pregnancy: a randomized, double-blind study controlled with placebo[J]. J Hepatol, 1997, 27 (6): 1022-1028.
41Williamson C, Hems LM, Goulis DG, et al. Clinical outcome in a series of cases of obstetric cholestasis identified via a patient support group[J]. BJOG, 2004, 111 (7): 676-681.
42Kenyon AP, Piercy CN, Girling J, et al. Obstetric cholestasis, outcome with active management: a series of 70 cases[J]. BJOG, 2002, 109 (3): 282-288.
43Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management[J]. Eur J Obstet Gynecol Reprod Biol, 2002, 100 (2): 167-170.
44Kremer AE, Beuers U, Oude-Elferink RP, et al. Pathogenesis and treatment of pruritus in cholestasis[J]. Drugs, 2008, 68 (15): 2163-2182.
45Bolier R, Oude Elferink RP, Beuers U. Advances in pathogenesis and treatment of pruritus[J]. Clin Liver Dis, 2013, 17 (2): 319-329.
46Kuiper EM, van Erpecum KJ, Beuers U, et al. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: results of a double-blind, randomized, placebo-controlled trial[J]. Hepatology, 2010, 52 (4): 1334-1340.
47Krause K, Kessler B, Weller K, et al. German version of ItchyQoL: validation and initial clinical findings[J]. Acta Derm Venereol, 2013, 93 (5): 562-568.
48Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy[J]. Gastroenterology, 2005, 129 (3): 894-901.
49Khurana S, Singh P. Rifampin is safe for treatment of pruritus due to chronic cholestasis: a meta-analysis of prospective randomizedcontrolled trials[J]. Liver Int, 2006, 26 (8): 943-948.
50Tandon P, Rowe BH, Vandermeer B, et al. The efficacy and safety of bile Acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus[J]. Am J Gastroenterol, 2007, 102 (7): 1528-1536.
51Mayo MJ, Handem I, Saldana S, et al. Sertraline as a first-line treatment for cholestatic pruritus[J]. Hepatology, 2007, 45 (3): 666-674.
52Jones DE. Fatigue in cholestatic liver disease: is it all in the mind?[J]. J Hepatol, 2007, 46 (6): 992-994.
53Abbas G, Jorgensen RA, Lindor KD. Fatigue in primary biliary cirrhosis[J]. Nat Rev Gastroenterol Hepatol, 2010, 7 (6): 313-319.
54Jones DE, Newton JL. An open study of modafinil for the treatment of daytime somnolence and fatigue in primary biliary cirrhosis[J]. Aliment Pharmacol Ther, 2007, 25 (4): 471-476.
55Boone RH, Cheung AM, Girlan LM, et al. Osteoporosis in primary biliary cirrhosis: a randomized trial of the efficacy and feasibility of estrogen/progestin[J]. Dig Dis Sci, 2006, 51 (6): 1103-1112.
57Levy C, Harnois DM, Angulo P, et al. Raloxifene improves bone mass in osteopenic women with primary biliary cirrhosis: results of a pilot study[J]. Liver Int, 2005, 25 (1): 117-121.
58Newton J, Francis R, Prince M, et al. Osteoporosis in primary biliary cirrhosis revisited[J]. Gut, 2001, 49 (2): 282-287.
(2015-12-28收稿)
DOI:10.3969/j.issn.1008-7125.2016.01.009
*本文通信作者,陆伦根,Email: lungenlu1965@163.com
猜你喜欢
小学生学习指导(当代教科研)(2021年6期)2021-05-23
马克思主义哲学研究(2020年1期)2020-11-26
人大建设(2019年12期)2019-11-18
中国现代医生(2016年23期)2016-11-15
科技视界(2016年21期)2016-10-17
中国实用医药(2016年24期)2016-10-17
中国实用医药(2016年24期)2016-10-17
中国实用医药(2016年24期)2016-10-17
人生十六七(2015年3期)2015-02-28
