
不依赖于剪接位点信号的高精度转录组序列比对算法①
2016-02-20 06:52张勇,徐云
计算机系统应用 2016年12期
张 勇, 徐 云
1(中国科学技术大学 计算机科学与技术学院, 合肥 230027)2(中国科学技术大学 安徽省高性能计算重点实验室, 合肥 230027)3(国防科学技术大学 高性能计算协同创新中心, 长沙 410073)
不依赖于剪接位点信号的高精度转录组序列比对算法①
张 勇1,2, 徐 云1,3
1(中国科学技术大学 计算机科学与技术学院, 合肥 230027)2(中国科学技术大学 安徽省高性能计算重点实验室, 合肥 230027)3(国防科学技术大学 高性能计算协同创新中心, 长沙 410073)
高通量转录组测序技术已经发展成为分析不同细胞中选择性剪接事件的最有效方法, 其测序数据处理的第一步是将数以百万的测序片段准确地比对到参考序列上, 称之为转录组序列比对. 现有的比对工具基本上都是依赖于经典的剪接位点信号, 一定程度上限制了转录组测序技术发现全新剪接位点的能力. 为此, 我们设计了一种不依赖于剪接位点信号的转录组序列比对方法RNAMap, 该方法按照重叠种子方式划分测序片段, 使用带有左右锚点的窗口扫描参考序列, 找出种子中含有的剪接位点. 计算实验表明, RNAMap精确度高达95%, 召回率也明显优于其他算法.
选择性剪接; 高通量转录组测序; 滑动窗口; 剪接位点
真核生物的基因是断裂基因, 由内含子序列和外显子序列组成, 选择性剪接是一种重要的转录后修饰过程, 在此期间, 前体RNA中的一个或多个内含子片段被剪切除去, 然后剩余的外显子拼接称为成熟的mRNA, 如图1. 选择性剪接使得基因能够产生多样的转录本, 而且人类基因组中90%以上的多外显子基因会发生选择性剪接. 相关研究表明, RNA剪接发生异常与人类的许多疾病密切相关[1].
定性和定量研究转录组的传统方法是构建cDNA或表达序列标签(EST)文库, 然后通过Sanger测序进行后续分析. 但是, 因为Sanger测序技术的成本较高且通量较低, 所以这种方法十分昂贵和低效. 随着下一代测序技术(next-generation sequencing, NGS)的迅猛发展, 高通量转录组测序(RNA-seq)在分析全基因组的剪接信息, 尤其是选择性剪接事件方面展现出了极佳的性能, 并已经发展成为研究剪接转录本的最有效的技术[2]. 目前, RNA-seq已能够应用于疾病的临床诊断; 此外, 在基础生物学研究中RNA-seq也有广泛的应用, 如分析不同的基因在不同阶段的表达情况.

图1 真核细胞基因结构图
RNA-seq分析软件的一项重要功能便是重建剪接之前的mRNA在细胞中的形态, 此外, 还应该能够评估每一种剪接异构体的表达水平. 然而, 所有分析过程的第一步都是要将RNA-seq中得到的测序片段(reads)比对到基因组上的原始位置, 而这些短片段的长度从数十碱基到数百碱基不等, 数量有几十万甚至几百万和上千万, 所以, 比对的过程是极其耗时的.
事实上, 如果测序片段完全来自于外显子序列,那么常规的序列比对工具(BWA[3]、Bowtie[4]等)便可以应对这种比对工作. 但是, 有大量的短片段是来自于两个甚至多个外显子序列, 在人类基因组中两个外显子序列一般间距20bp~500000bp, 这远远超过了常规序列比对工具处理的范围. 因此, 研究的主要问题便是如何将跨越剪接位点的测序片段快速且准确地比对到参考序列上.
为了解决上述问题, 早期的策略是根据已有的基因组注释文件, 利用常规序列比对工具将测序片段定位到基因组上. 虽然这种方法可以定位大部分的测序片段, 但它的局限性也不容忽视. 毕竟, 即使是目前人们研究的最为深入的人类基因组, 它的注释文件仍然是不完整的, 所以上述策略是无法识别未在注释文件中出现的全新的剪接位点, 而这也就使RNA-seq丧失了发现新剪接异构体的能力.
事实上, 近年来也相继出现了一些不依赖于基因组注释文件的RNA-seq序列比对工具, 比如SpliceMap、MapSplice、TopHat[5]、CRAC[6]、 OLego[7]和HISAT[8]等. 其中, TopHat系列软件是最具有代表性,也是目前使用最广的比对工具. 它采用外显子优先的策略, 整个比对过程分为两个阶段. 第一阶段, 利用Bowtie将测序片段定位到参考基因组上, 这样, 含有剪接位点的测序片段就会被过滤出来; 然后通过MAQ中的组装模块将成功定位的短片段组装起来.经过这一阶段, 供体位点和受体位点的侧翼序列拼接起来组成潜在的剪接序列, 作为下一阶段的参考序列.第二阶段, 将在第一阶段中未成功定位的测序片段比对到上述由外显子拼接成的序列上. 然而, TopHat在拼接外显子序列时仅仅考虑经典的剪接位点(GT/C-AG), 虽然目前已知的具有经典信号的剪接位点占了绝大多数, 但是有研究表明非经典剪接位点的比例很有可能被低估了[9,10]. 因此, TopHat存在的主要问题是会遗漏具有非经典剪接信号的测序片段. 其余几个RNA-seq序列比对软件虽然分别采用了各自不同的比对策略, 但在默认情况下也都是依赖了经典的剪接信号, 所以也具有与TopHat类似的缺陷. 尽管个别软件可以通过设定参数来穷尽所有类型的剪接位点信号, 但是算法的复杂度较高. 总之, 目前依赖于经典剪接位点信号的比对算法已经发展的较为完善, 但尚缺乏对不依赖于剪接信号的比对算法的研究.
为了克服以上不足, 我们使用带有左右锚点的窗口扫描参考序列, 设计了命名为RNAMap的转录组序列比对工具.
1 方法
RNAMap的执行过程分为两个阶段. 第一阶段, RNAMap尝试利用常规的序列比对工具将原始数据集中的所有的测序片段定位到参考基因组上. 在这一阶段中, 完全来自于一个外显子序列的测序片段可以被直接比对到基因组上, 这样没有比对上的测序片段就可能含有剪接位点. 第二阶段, 对于这些未比对上的片段, 利用两个表来寻找其中的剪接位点. RNAMap的执行流程如图2所示. RNAMap将测序片段划分成几个重叠的种子, 每个种子可发现一个剪接位点, 这样我们的方法就能够处理含有多个剪接位点的测序片段.
1.1 初始比对阶段
RNAMap使用Bowtie来处理RNA-seq的reads,将它们比对到参考序列上. 如果存在基因组注释文件(文件中记录了原基因组中外显子序列的位置), 那么可以此文件为基础生成转录组序列(只包含外显子序列), 并将其作为参考序列. 采用这种策略, 一方面可以提高序列比对的敏感性和准确性; 另一方面也可以加速比对的过程. 如果无法获得有效的注释文件, 那么RNAMap会选择基因组作为参考序列.
即使以转录组作为参考序列, 也仍然会有一些reads无法成功地定位到参考序列上, 可能因为这些reads中被错误测序的碱基数超出了RNAMap设定的阈值, 另一个重要的原因是产生这些短片段的转录本信息并没有记录在注释文件中. 此外, 由于假基因的存在[11], 也会有一些短片段被错误的定位到参考序列上.

图2 RNAMap流程图
1.2 分段比对阶段
一个剪接位点可以将一个read分成两个片段(segments), 但事实上, 这些segments并不是完全随机的分布在基因组上. 如果我们不考虑一些特殊的情况,比如基因融合, 剪接位点分割一个read产生的segments应该被定位到同一个染色体上, 并且满足一定的距离限制, 对于人类及其他哺乳动物, 一般为20bp~500000bp. 如果先分别独立的定位这些segments, 然后再根据位置限制条件进行过滤会, 那么这样会增加搜索空间. 因此, 如果在比对segments的同时添加有一定的限制条件, 那么既可以减小搜索空间, 又可以精简后续的筛选过程. 正是基于这样的考虑, 分段比对阶段分为以下三个步骤.
1.2.1 划分种子
将测序得到的片段划分成互相重叠的种子, 例如,将长度为100bp的测序片段划分为三个长为50bp的种子, 它们在原测序片段上的区间分别为[1,50]、[26,75]和[51,100]. 转录组测序深度可以保证每一个剪接位点至少会被一个种子所覆盖.
1.2.2 种子定位
上一步产生的种子可以分为两类: 一类种子不含有剪接位点; 另一类种子含有剪接位点, 并且我们假设它们仅含有一个剪接位点. RNAMap调用Bowtie来比对所有的种子, 第一类种子可以被成功地定位到参考序列上, 第二类种子被过滤出来, 然后建立两个表进行索引, 一个为静态表, 另一个为动态表. 此外, 种子中的剪接位点既可能出现在种子的前半段, 也可能出现在后半段. 下面仅讨论剪接位点出现在后半段的情况, 以种子的前半段序列作为左锚点, 后1/4序列作为右锚点; 至于另外一种情况, 可以用一种对称的方法来实现. 为了能够处理含有误配的情况, RNAMap采用了与PerM[12]类似的单周期空间种子的方法.
① 静态表
以种子的前半段(左锚点)作为键, 以种子的标识号作为值, 建立静态表. 所有种子的键-值对信息都需要加入到表中, 并且静态表一旦建立, 在比对的过程中都将保持不变, 如图3.
② 动态表
动态表用于存储右锚点序列与左锚点比对信息组成的键-值对. 有两个窗口沿着参考基因组进行滑动,窗口S的长度与静态表键的长度相等, 用来查询静态表; 窗口D的长度一般为种子长度的1/4, 用来查询动态表.

图3 静态表示例
当窗口S中的序列在静态表中查询到时, 表明该种子的前半段可以定位到此处, 之后继续向后延伸比对, 直至遇到第一个无法匹配的位点, 最后以该种子的后1/4片段为键, 以前面的定位信息(包括标志号、定位起点、比对的碱基数)为值, 插入动态表.
当窗口D中的序列在动态表中查询到时, 表明该种子的后1/4片段可以定位到此处, 之后继续向前延伸比对, 直至遇到第一个无法匹配的位点, 最后结合动态表中对应的值中保存的定位信息, 就可以判定种子是否能够分段比对到参考基因组上的两个位置. 如果前后两部分比对的位置超出了预设的距离范围, 则需要将动态表中对应的键-值信息删除. 此外, 每当扫描完一个染色体的序列, 也需要将动态表中的信息全部清空, 以保证种子的两部分定位到同一个染色体上.
1.2.3 种子整合
将种子的定位结果组合成reads的定位信息. 在这一过程中需要检查种子比对位置的一致性, 从而将符合要求的种子组合成完整的reads.
2 实验结果
我们分别在模拟数据集和真实数据集上测试RNAMap的性能, 并与其他主流软件进行对比. 为了保证实验的可靠性和公平性, 所有的软件都在同一台计算机上运行, 其基本的配置为Intel(R) Core(TM) i7-4770K CPU, 24G RAM, 64-bit Ubuntu 14.04 OS.
2.1 模拟数据集测试
我们使用FluxSimulator[13]软件, 以人类基因组GRCh38及其注释文件为基础, 随机模拟产生了1000000条长为100bp的测序片段(reads), 每条序列的来源信息保存在BED格式的文件中, 因此我们可以计算各软件比对结果的召回率(Recall Rate)和精确度(Precision), 结果如表1.
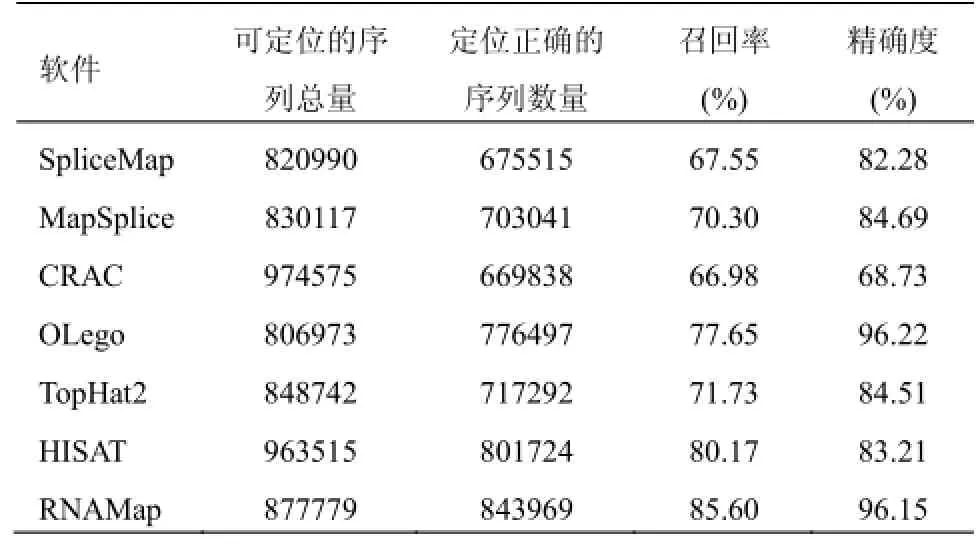
表1 各软件的模拟数据集比对结果统计
由表1可知, 虽然CRAC和HISAT可以将绝大多数的测序片段(分别为97.46%和96.36%)比对到基因组上, 但是精确度比较低, 尤其是CRAC的精确度不足70%. TopHat2、SpliceMap、OLego、MapSplice、CRAC的召回率都比较低, 虽然HISAT的召回率达到80.17%,但是它的精确度也仅仅高于SpliceMap和CRAC. 因为我们的模拟数据集是随机产生的, 所以其中有大量的reads含有非经典的剪接位点, 但上述软件无法处理此类情况, 从而造成其比对的质量相对较低.
RNAMap的精确度高达96.15%, 虽然稍低于OLego的96.22%, 但是RNAMap的召回率达到了85.60%, 明显高于其它几个软件. 这是因为在比对的整个过程中, RNAMap并未受经典剪接位点信号的限制, 因而可以更准确地将各类reads比对到参考序列上.
2.1 真实数据集测试
我们在73685727条长为100bp的真实测序片段数据集(来源于K562细胞系, 是一种人类的白血病细胞, GEO序列号为GSM1838573)上比较各个软件的性能,结果如图4所示.
由图4可知, RNAMap可以将68647397条测序片段(93.16%)定位到参考序列上, 明显优于TopHat2、SpliceMap、OLego和MapSplice, 仅次于CRAC和HISAT. K562细胞系是一种癌变的细胞, 其选择性剪接事件也与正常细胞不同, 因此其测序得到的reads中会含有更多类型的剪接位点. 虽然我们无法统计真实数据集中正确的匹配位置, 但是根据模拟数据集的结果, 我们知道RNAMap的精确度在95%以上, 因此可以推断RNAMap的整体性能较佳.

图4 各软件的真实数据集比对结果统计
3 结语
本文针对高通量转录组测序的序列比对问题, 提出了一种使用带锚点的滑动窗口扫描参考序列的比对方法, 通过在模拟数据集和真实数据集上对算法的性能进行测试, RNAMap无论是在召回率, 还是在精确度和片段匹配率上都表现出较优的性能. 虽然在测序片段为100bp的大小为1000000的数据集上, RNAMap比目前最快的软件HISAT大约多耗时40%, 但是时间仍然在可接受的范围内, 而且可以获得更高的精确度.该方法不借助经典的剪接位点信号, 因此可以充分发挥RNA-seq的优势, 识别基因组注释文件中没有记录的全新的剪接位点. 接下来的工作, 一是需要提高RNAMap的比对速度; 二是解决含有多个剪接位点的种子的比对问题, 这样可以降低对测序深度和种子重叠度的要求.
1 Nagao K, Togawa N, Fujii K, et al. Detecting tissue-specific alternative splicing and disease-associated aberrant splicing of the PTCH gene with exon junction microarrays. Human Molecular Genetics, 2005, 14(22): 3379–3388.
2 Mcgettigan PA. Transcriptomics in the RNA-seq era. Current Opinion in Chemical Biology, 2013, 17(1): 4–11.
3 Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25(14): 1754–1760.
4 Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology, 2009, 10(3): R25.
5 Garber M, Grabherr MG, Guttman M, et al. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods, 2011, 8(6): 469–477.
6 Philippe N, Salson M, Commes T, et al. CRAC: An integrated approach to the analysis of RNA-seq reads. Genome Biology, 2013, 14(3): R30.
7 Wu J, Anczukow O, Krainer AR, et al. OLego: Fast and sensitive mapping of spliced mRNA-seq reads using small seeds. Nucleic Acids Research, 2013, 41(10): 5149–5163.
8 Kim D, Langmead B, Salzberg SL. HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 2015, 12(4): 357–360.
9 Filichkin SA, Priest HD, Givan SA, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Research, 2010, 20(1): 45–58.
10 Parada GE, Munita R, Cerda CA, et al. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Research, 2014, 42(16): 10564–10578.
11 Kalyana-Sundaram S, Kumar-Sinha C, Shankar S, et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell, 2012, 149(7): 1622–1634.
12 Chen Y, Souaiaia T, Chen T. PerM: Efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. Bioinformatics, 2009, 25(19): 2514–2521.
13 Griebel T, Zacher B, et al. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Research, 2012, 40(20): 10073–10083.
Highly Precise Transcriptome Sequence Alignment Algorithm Independent From Splice Site Signals
ZHANG Yong1,2, XU Yun1,312
(School of Computer Science and Technology, University of Science and Technology of China, Hefei 230027, China) (Key Laboratory of High Performance Computing of Anhui Province, University of Science and Technology of China, Hefei 230027, China)3(Collaborative Innovation Center of High Performance Computing, National University of Defense Technology, Changsha 410073, China)
RNA-seq has become the most effective method of analyzing alternative splicing events in different types of cells. The first step of processing data of RNA-seq is to exactly align millions of sequencing fragments against the reference sequence, which is called transcriptome sequence alignment. The existing sequence alignment tools for RNA-seq almost rely on canonical splice site signals, which, to some extent, limits the ability to identify novel splice sites. Therefore, we design a method independent from splice site signals, named RNAMap. It divides the sequencing fragments according to overlapping seeds method and scans the reference sequence via sliding windows with left and right anchors. In this way, splice sites within seeds can be identified. The computational experiments indicate that RNAMap not only reaches a precision of over 95%, but also outperforms the existing softwares in recall rate.
alternative splicing; RNA-seq; sliding windows; splice sites
国家自然科学基金(60533020)
2016-03-17;收到修改稿时间:2016-04-11
10.15888/j.cnki.csa.005443
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
——《艺术史导论》评介
美育学刊(2022年5期)2022-10-18
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
电子技术与软件工程(2020年22期)2021-01-30
数字技术与应用(2020年12期)2021-01-22
中国生殖健康(2020年4期)2021-01-18
移动通信(2020年5期)2020-06-08
山东煤炭科技(2020年1期)2020-03-06
新教育时代·教师版(2017年30期)2017-09-12
湖北农业科学(2014年11期)2014-09-10
