
全自动固相萃取液质联用法测定动物源性食品中镇静剂类药物残留
2016-02-17 03:12:06朱群英索莉莉朱玉玲
食品工业科技 2016年24期
朱群英,索莉莉,朱玉玲,李 良
(南昌市疾病预防控制中心,江南南昌 330038)
朱群英,索莉莉,朱玉玲,李 良
(南昌市疾病预防控制中心,江南南昌 330038)
建立全自动固相萃取-液质联用同位素内标法测定动物源性食品中13种镇静剂的方法。样品中添加同位素内标地西泮-d5和氯丙嗪-d6,经乙腈提取,全自动固相萃取仪净化,Waters ACQUITY UPLCTMBEH C18(2.1 mm×100 mm,1.7 μm)不锈钢色谱柱分离,三重四级杆质谱多反应监测方式检测,同位素内标法定量。13种镇静剂在线性范围内具有良好的线性关系,相关系数r≥0.999,检出限在0.030~0.113 μg/kg之间,加标回收率在88.5%~115.0%之间,相对标准偏差在2.45%~7.34%之间。该方法样品前处理简单,净化过程实现了自动化操作,大大提高了工作效率;该方法灵敏度高、回收率好,适用于动物源性食品中多种镇静剂的同时检测。
全自动固相萃取,液质联用,动物源性食品,镇静剂
镇静剂(sedative)是指能使中枢神经系统产生轻度抑制,减弱机能活动,从而起到消除躁动不安、恢复安静的一类药物。近年来,个别畜牧业饲养者因经济利益的驱使,擅自在畜禽饲养过程中添加镇静剂类药物以起到镇静催眠、增重催肥、缩短出栏时间的作用;另外,在动物运输过程中,为减少动物死亡和体重下降,防止肉品质降低,也常使用镇静剂类药物以减少应激带来的损失。非法使用此类药物会使其原形和代谢产物不可避免地残留于动物源食品中,人们食用了这些食品后会对人体中枢神经系统等造成不良影响,因此许多国家都将此类药物列为禁用药物。
目前,国内外报道的镇静剂类药物残留检测的仪器方法主要有液相色谱法(LC)[1-2]、气相色谱-质谱联用法(GC-MS)[3-4]和液相色谱-串联质谱联用法(LC-MS/MS)[5-14]等。其中LC法灵敏度较低,无法满足对禁用药物检测的要求。GC-MS有较好的灵敏度,但部分药物的前处理需衍生化费时较长。LC-MS/MS作为目前报道较多的方法,可以进行多残留定性定量检测。由于动物源性样品基质复杂,为不干扰检测,提取后的样品需采用固相萃取(SPE)[1-6,13]、液一液萃取(LLE)[8,10]、分散固相萃取(QuEChERS)[14]等方法对样品进行净化,这些前处理方法不能自动化操作,不利于大批量样品的快速检测。全自动固相萃取法可自动完成活化、加样、洗脱、收集等全过程,可以节省时间、减少交叉污染、重现性好。本研究采用全自动固相萃取与超高效液相色谱/质谱联用技术同时检测动物源性食品中的苯二氮卓类和吩噻嗪类药物残留。
1 材料与方法
1.1 材料与仪器
甲醇、甲酸 为LCMS级;乙腈 为色谱纯;三氯乙酸、氨水 为分析纯;超纯水;标准物质:硝西泮(Nitrazepam)、氯硝西泮(Clonazepan)、地西泮(Diazepam)、氟硝西泮(Flunitrazepam)、奥沙西泮(Oxazepam)、劳拉西泮(Lorazepam)、阿普唑仑(Alprazolam)、替马西泮(Temazepam)等8种苯二氮卓类混合标准溶液,浓度为250μg/mL,地西泮-d5(Diazepam-d5)内标,浓度为1.0 mg/Ml 以上标准购于SIGMA-ALDRICH;异丙嗪(Promethazine hydrochloride)、甲苯噻嗪(Xylazine hydrochloride)、乙酰丙嗪(Acepromazine maleate salt)、丙酰丙嗪(propionylpromazine hydrochloride)、氯丙嗪(chlorpromazine hydrochloride)、氯丙嗪-d6(chlorpromazine-d6)内标,纯度>98% 购于Dr.Ehrenstorfer公司。
超高效液相色谱仪LC-30A与三重四级杆质谱仪LCMS-8040联用系统 日本岛津公司:配置电喷雾离子源(ESI),Peak Scientific氮气发生器(NM32L),LC-30A×2输液泵,DUG-20A5在线脱气机,SIL-30AC自动进样器,CTO-30AC柱温箱,CBM-20A系统控制器,LCMS-8040三重四级杆质谱仪,LabSolutions色谱工作站;GX274自动固相萃取仪 美国吉尔森;Milli-Q Direct8 超纯水机 默克密理博;5424台式高速离心机 德国eppendorf;N-EVNP24氮吹仪 美国Organomation。
1.2 实验方法
1.2.1 液相色谱条件 Waters ACQUITY UPLCTMBEH C18(2.1×100 mm,1.7 μm)不锈钢色谱柱;柱温:40 ℃;进样体积:5 μL;流速:0.4 mL/min;流动相:A为0.1% 甲酸水,B为甲醇,梯度洗脱程序:0~2 min,30%的B流动相;2~2.2 min,30%~50%,维持4.8 min;7~12 min,50%~80%的B流动相;12~13 min,80%~100%的B流动相;维持3 min;16~17 min,100%~30%的B流动相,维持3 min。
1.2.2 质谱条件 离子源:电喷雾离子源ESI(+);离子源接口电压:4.5 kV;雾化气流量:氮气3.0 L/min;干燥气流量:15 L/min;碰撞气:氩气;脱溶剂管温度:250 ℃;加热模块温度:400 ℃;扫描模式:多反应监测MRM;驻留时间:50 ms;延迟时间:1 ms。
1.2.3 全自动固相萃取条件 固相萃取柱:Oasis HLB柱,3 mL;吸样品速度:3 mL/min;吸溶剂速度:5 mL/min;吸气泡速度:0.3 mL/min;排样品速度:2 mL/min;排溶剂速度:5 mL/min;排气泡速度:0.3 mL/min。
Oasis HLB固相萃取柱依次用5 mL甲醇、5 mL水活化,将样品提取液5 mL全部通过,依次用5 mL水、3 mL的15%乙腈水淋洗后抽干,5 mL 5%乙酸甲醇洗脱并接收,洗脱液用氮气吹干后定容待测。
1.2.4 标准溶液的配制 精密称取异丙嗪、甲苯噻嗪、乙酰丙嗪、丙酰丙嗪、氯丙嗪2.50 mg,用甲醇稀释定容到10 m L,得到250 μg/mL的噻吩嗪类混合标准储备溶液。分别取40 μL苯二氮卓类混合标准溶液250 μg/mL和40 μL噻吩嗪类混合标准储备溶液,用甲醇稀释定容到10 mL,得到13种镇静剂混合标准溶液,浓度为1 μg/mL。再稀释成0.05 μg/mL的混合标准使用液。
精密称取氯丙嗪-d6内标2.50 mg,用甲醇稀释定容到10 mL,浓度为 250 μg/mL。分别取250 μg/mL氯丙嗪-d6 40 μL和 1.0 mg/mL地西泮-d5 10 μL,用甲醇稀释定容到10 mL,得到2种镇静剂混合内标溶液,浓度为1 μg/mL。再稀释成0.05 μg/mL的内标使用液。
1.2.5 样品溶液的制备 准确称取2.00 g(精确到0.01 g)样品于50 mL离心管中,加入10 mL乙腈,再加入0.05 μg/mL的混合内标溶液200 μL,漩涡1 min,超声提取10 min。以5000 r/min的转速下离心10 min,取上清液5 mL,在50 ℃下用氮气吹至近干,用20%氯化钠溶液定容到5 mL,经全自动固相萃取仪净化洗脱后,洗脱液用氮气吹干,残渣用初始流动相溶解,涡旋混合1 min,定容到1 mL,过0.22 μm微孔滤膜,准备上机。
2 结果与讨论
2.1 液相色谱条件的优化
2.1.1 色谱柱的选择 实验比较了两个不同规格的色谱柱1:Waters ACQUITY UPLCTMBEH C18(2.1 mm×100 mm,1.7 μm)不锈钢色谱柱,2:Waters ACQUITY UPLCTMHSSC18(2.1 mm×100 mm,1.7 μm)不锈钢色谱柱,在相同的流动相和流速下,柱1的离子化效率高,且分离效果好,所以选择柱1为本实验用的分析柱。
2.1.2 流动相的优化 实验分别采用了0.1%甲酸水和甲醇、0.1%甲酸水和乙腈、0.01 mol/L乙酸铵和甲醇、0.01 mol/L乙酸铵和乙腈等四组流动相,经比较用0.1%甲酸水和甲醇为流动相时,各组分离子化效率高,峰型好,干扰少,本实验采用0.1%甲酸水和甲醇为流动相;由于这13种镇静剂的化学性质差异较大,在柱子上的保留不同,实验采用梯度洗脱。
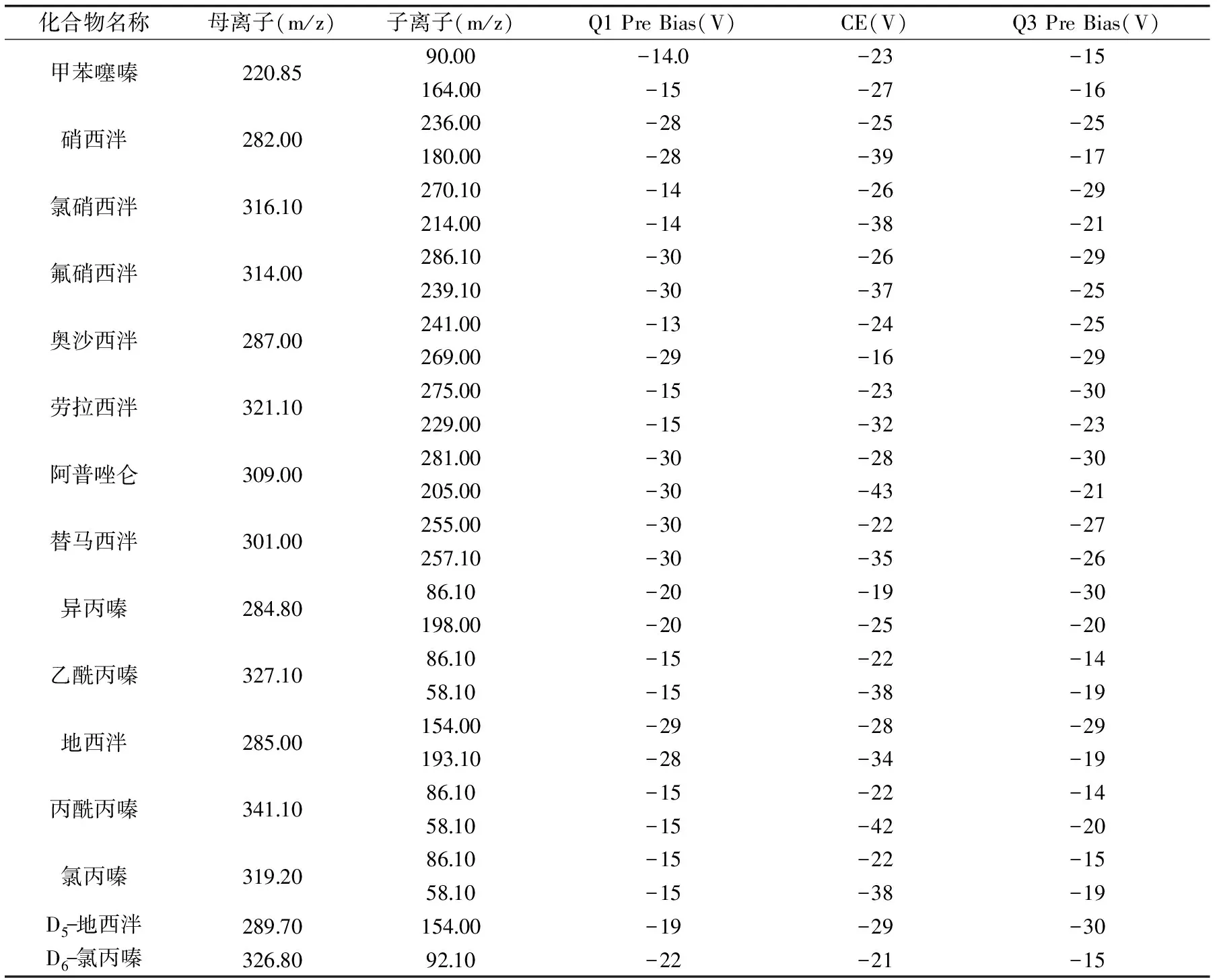
表1 13种镇静剂和2种内标物的MRM参数
2.2 质谱条件的优化
2.2.1 母离子的优化 苯二氮卓类镇静剂药物属于中性或弱碱性药物,噻吩嗪类镇静剂药物也是弱碱性药物,他们均更适合在ESI源的正离子模式下离子化,产生的母离子为[M+H]+,峰响应高。将1 μg/mL的各镇静剂标准溶液,在不接色谱柱的情况下进样1 μL,ESI(+)模式下进行Q3 Scan,确定各镇静剂的母离子质荷比。
2.2.2 子离子的选择以及碰撞能的优化 在优化好的母离子情况下,将1 μg/mL的各镇静剂标准溶液,在不接色谱柱的情况下,选取响应较高的两对子离子,同时对Q1 Pre Bias电压、碰撞能量、Q3 Pre Bias电压进行优化,优化好的子离子以响应值最高的为定量离子,响应其次的为定性离子。MRM参数见表1,其多反应监测(MRM)见图1。
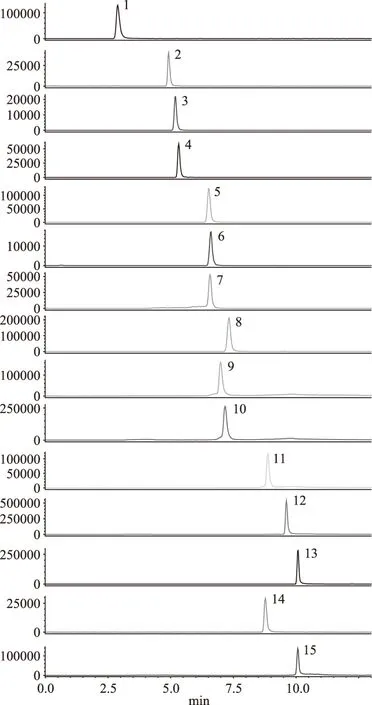
图1 13种镇静剂标准品及2种同位素内标物的多反应监测(MRM)色谱图Fig.1 The MRM chromatogram of 13 kinds of sedative standards and 2 kinds of isotope internal standards注:1:甲苯噻嗪;2:硝西泮;3:氯硝西泮;4:氟硝西泮;5:奥沙西泮;6:劳拉西泮;7:阿普唑仑;8:替马西泮;9:异丙嗪;10:乙酰丙嗪;11:地西泮;12:丙酰丙嗪;13:氯丙嗪;14:地西泮-d5;15:氯丙嗪-d6。
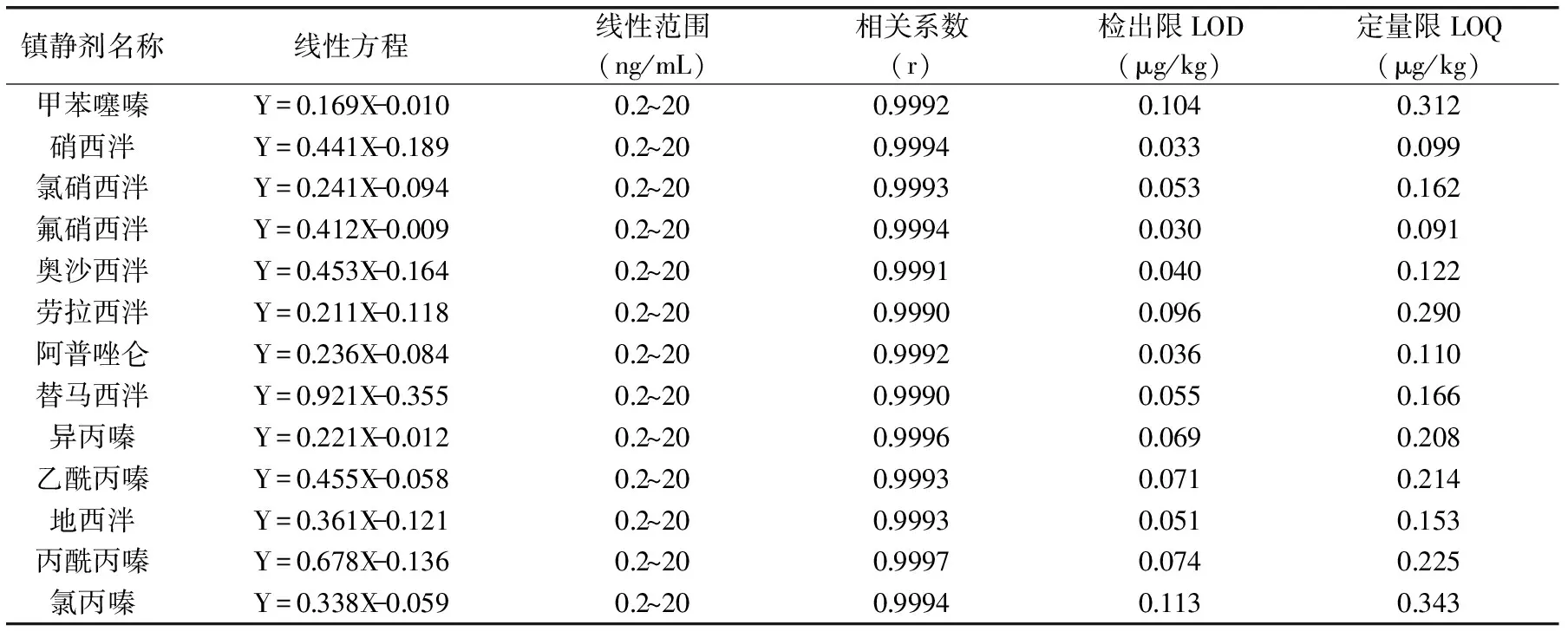
表2 13种镇静剂的线性方程、线性范围、相关系数、检出限与定量下限
2.3 全自动固相萃取条件的优化
2.3.1 固相萃取柱的选择 对于固相萃取实验,固相萃取柱的选择至关重要,分别采用HLB柱、C18柱和 MCX柱进行实验,结果发现C18柱对苯二氮卓类镇静剂药物有较高的回收,但对噻吩嗪类镇静剂药物回收率低;MCX柱对噻吩嗪类镇静剂药物回收率较高,但对苯二氮卓类镇静剂药物回收率低;HLB柱对这两类13种药物残留均有较好的回收率,故选HLB柱为本实验的固相萃取柱。
2.3.2 全自动固相萃取仪参数的优化 由于样品溶液粘度较大,吸样速度过快会产生气泡,定量不准确,吸样速度慢又影响工作效率,我们对吸样速度进行优化,结果表明当吸样速度为3 mL/min时,不会产生气泡,故选3 mL/min为吸样速度;同时为了让样品能更好的保留在固相萃取柱中,我们对排样速度进行优化,为2 mL/min。其他全自动固相萃取仪参数参考厂家推荐的参数进行实验。
为了减少干扰,需采用尽可能大比例的有机相除杂,但有机相比例过大,被测物也会被洗脱下来。分别采用了3 mL的10%乙腈水、15%乙腈水、20%乙腈水实验,实验结果表明,当使用20%乙腈水除杂时,替马西泮在洗脱液中被检出,15%乙腈水洗脱液中未发现分析物,故选用3 mL的15%乙腈水为除杂溶剂。
分别采用2%乙酸甲醇、5%乙酸甲醇为洗脱溶剂洗脱样品,当使用2%乙酸甲醇为洗脱溶剂时,苯二氮卓类化合物回收率高,但吩噻嗪类化合物回收率低,当使用2%乙酸甲醇为洗脱溶剂时13种化合物都有较好的回收率,故选用5 mL 5%乙酸甲醇为洗脱溶剂。
2.4 方法学实验
2.4.1 标准曲线及方法的检出限和定量限 用移液器吸取混标溶液和内标溶液适量,用甲醇0.1%甲酸水溶液(30∶70,V∶V)配成含各镇静剂0.2、0.5、1.0、2.0、5.0、10.0、20.0 ng/mL的标准系列,每份溶液含同位素内标5 ng/mL,其中苯二氮卓类以d5-地西泮为内标计算,噻吩嗪类以d6-氯丙嗪为内标计算,以待测化合物和内标的峰面积比值为纵坐标,待测化合物的浓度为横坐标进行回归分析,得到线性回归方程、及相关系数(见表2)。
用空白猪瘦肉作为基质,添加不同量的镇静剂标准品,按1.2.5样品前处理方法进行处理后测定,以S/N=3为检出限,S/N=10为定量限(见表2)。
由表2可知,采用同位素内标法定量,各化合物的线性关系良好,相关系数r值均大于0.999。
2.4.2 方法的回收率和精密度 称取猪瘦肉空白样品,分别加入高中低三个浓度的各镇静剂的混合标准溶液,按1.2.5样品前处理方法进行处理,每个加标水平平行测定6次,计算13种镇静剂的回收率和相对标准偏差,结果见表3。
由表3可知,采用同位素内标法计算,13种镇静剂回收率在88.5%~115.0%之间。
2.4.3 稳定性实验 准确吸取高、中、低三个浓度的混合标准溶液,在最优化的色谱质谱条件下每隔1 h进样一次,连续6次,各组分峰面积的RSD在0.63%~3.49%之间;每隔5 d进样一次,连续6次,各组分峰面积的RSD在2.12%~7.33%之间;由此可知,在一个月内各标准溶液具有良好的稳定性。
2.4.4 样品检测 随机抽取了市场上的猪瘦肉、猪肝、猪肺、猪血、猪肾、牛肉、羊肉样品各10份进行检测,均未检测出镇静剂药物残留。分别取每类样品中的一个进行加标进行基质干扰实验,实验结果证明,猪肺基质干扰最大,但不影响样品的检测。
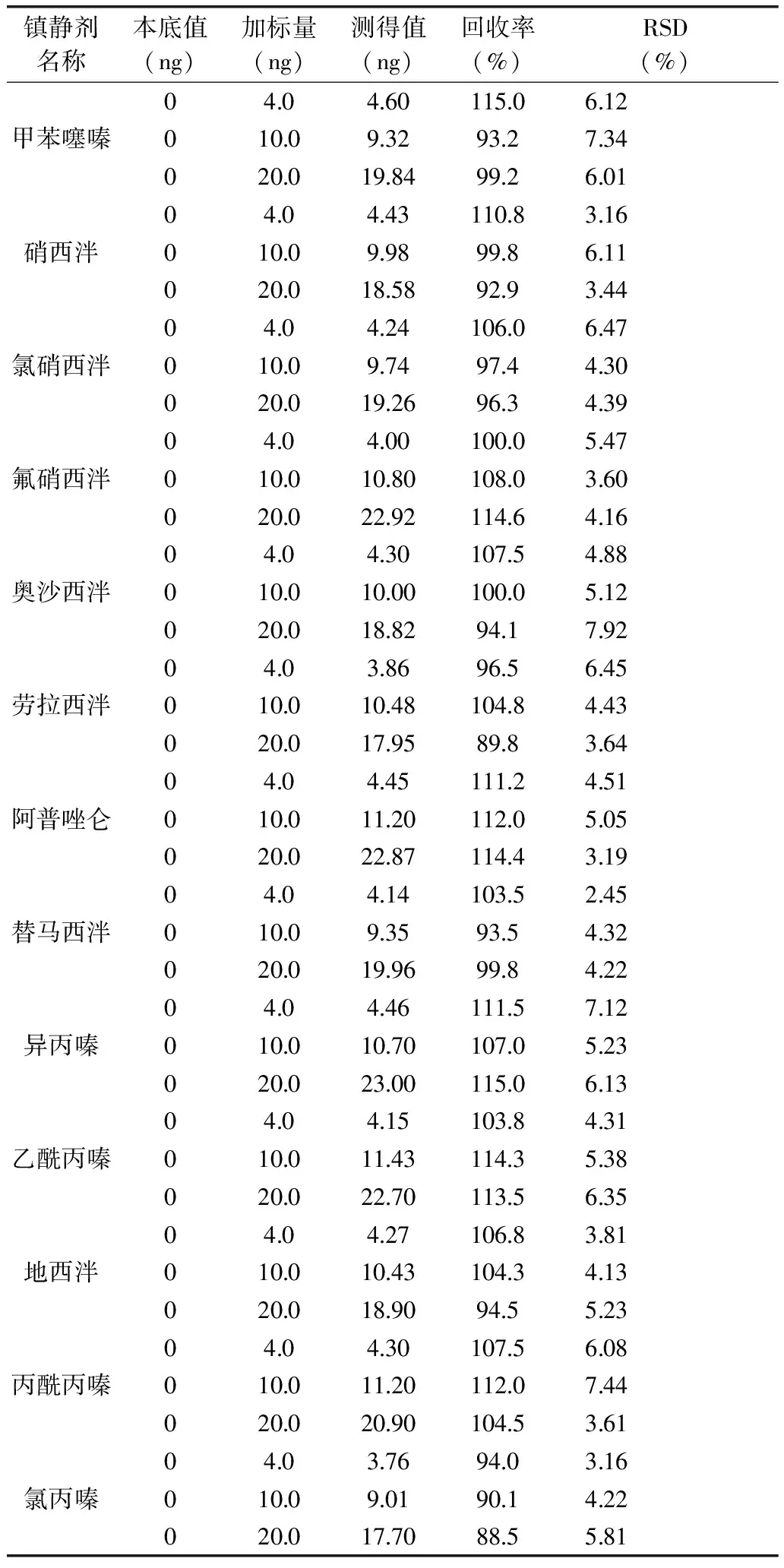
表3 13种镇静剂的回收率和精密度(n=6)
3 结论
本文采用同位素内标液质联用法同时测定动物源性食品中镇静剂类药物残留。采用全自动固相萃取仪对样品进行净化,实现样品前处理的自动化操作,可以提高工作效率。该方法样品前处理简单、交叉污染少;在线性范围内,有良好的线性相关性,回收率高,重现性好。三重四级杆液质联用多反应监测模式进行检测,同位素内标法定量,可以实现对组分多残留的同时检测。
[1]魏晋梅,罗玉柱,白云旭. 高效液相色谱法同时测定羊肉中的11种镇静剂类药物[J]. 食品工业科技,2014,35(10):95-97.
[2]Mohammad N. Validation of SPE-HPLC determination of 1,4-benzodianepines and metabolites in blood plasma,urine and saliva[J]. J SepSci,2008,31:3704-3717.
[3]谭贵良,赵天珍,王文林,等. 气相色谱-质谱法同时测定腊肠中7种镇静剂类药物残留[J]. 现代食品科技,2014,30(2):274-278.
[4]Linli Cheng,Yujie Zhang,Jianzhong Shen,et al. GC-MS method for simultaneous determination of four sedative hypnotic residues in swine tissues[J]. Chromatographia,2010(71):155-158.
[5]孙雷,张骊,徐倩,等. 超高效液相色谱-串联质谱法检测猪肉和猪肾种残留的10种镇静剂类药物[J]. 色谱,2010,28(1):38-42.
[6]钱晓东,于慧娟,惠芸华,等. 水产品中镇静剂残留的高效液相色谱-串联质谱法测定[J]. 湖南农业科学,2010(19):134-137.
[7]严丽娟,张洁,潘晨松,等. 超高效液相色谱-飞行时间质谱法高通量筛查乳制品中20种镇静剂[J]. 分析化学,2013,41(1):31-35.
[8]吴宁鹏,班付国,彭丽,等. 超高效液相色谱一串联四极杆飞行时间质谱法筛查饲料中11种镇静剂类药物[J]. 质谱学报,2012,33(2):94-99.
[9]Sooyeun Lee,Eunyoung Han,Sanghwan In,et al. Determination of illegally bused sedative-hypnotics in hair samples from drug offenders[J]. Journal of Analytical Toxicology,2011(35):312-315.
[10]BE Sminka,JE Brandsmaa,A Dijkhuizena,et al. Quantitative analysis of 33 benzodiazepines,metabolites and benzodiazepine-like substances in whole blood by liquid chromatography-(tandem)mass spectrometry[J]. Journal of Chromatography B,2004,811:13-20.
[11]EN Sauve,M Langdegard,D Ekeberg,et al. Determination of benzodiazepines in ante-mortem and post-mortem whole blood by solid-supported liquid-liquid extraction and UPLC-MS/MS[J]. Journal of Chromatography B,2012,883-884:177-188.
[12]Kristina Y Rust,Markus R Baumgartner,Natascha Meggiolaro,et al. Detection and validated quantification of 21 benzodiazepines and 3 “z-drugs”in human hair by LC-MS/MS[J]. Forensic Science International,2012,215:64-72.
[13]中华人民共和国国家质量监督检验检疫总局 中华人民共和国出入境检验检疫行业标准. SN/T 2113-2008进出口动物源性食品中镇静剂类药物残量的检测方法 液相色谱-质谱/质谱法[S]. 北京:中国标准出版社,2008.
[14]张毅,岳振峰,蓝芳,等. 分散固相萃取净化与液相色谱/串联质谱法测定牛奶中8类禁用药物残留[J]. 分析化学(FENXI HUAXUE)研究报告,2012,40(5):724-729.
Determination of sedative drug residues in animal derived food by automatic SPE combined with ultra performance liquid chromatography-tandem mass spectrometry
ZHU Qun-ying,SUO Li-li,ZHU Yu-ling,LI Liang
(Nanchang Center for Disease Prevetion and Control,Nanchang 330038,China)
A new approach for determination of 13 kinds of sedatives in animal derived food by automatic solid phase extration combined with ultra performance liquid chromatography-tandemmass spectrometry using isotope internal standard was described. The homogenized sample was spiked with diazepam-d5and chlorpromazine-d6,and extracted with acetonitrile,then purified by automatic solid phase extration. After the analytes were separated by Waters ACQUITY UPLCTMBEH C18(2.1 mm×100 mm,1.7 μm)column. All sedatives were detected by MS/MS system with eletrospray ion ization(ESI+)under multiple reaction monitoring(MRM)mode and quantified by the isotope internal standard technique. The result indicated that the LOD of 13 kinds of sedatives ranged from 0.030 μg/kg to 0.113 μg/kg. Meanwhile,the high correlation coefficents(r≥0.999)of 13 kinds of sedatives were obtained within their respective linear ranges. The average recoveries at low,intermediate and high spiked levels ranged from 88.5% to 115.0% with relative standard deviations(RSD)of 2.45%~7.34%. The method with simple sample preparation achieved the automated purification process which then improved considerably the work efficiency. This method was suitable for simultaneous determination of multiple sedatives in animal derived food with simple pretreatment,high sensitivity,good recovery.
automatic solid phase extration;LC-MS/MS;animal derived food;sedatives
2016-05-27
朱群英(1980-),女,硕士,主管技师;研究方向:食品理化检验,E-mail:zqy0797@163.com。
南昌市科技局(洪科发计字【2013】210号第61项)。
TS
A
1002-0306(2016)24-0000-00
10.13386/j.issn1002-0306.2016.24.000
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
快乐语文(2021年15期)2021-06-15 10:19:38
中国特种设备安全(2021年12期)2021-04-26 14:37:00
童话世界(2020年13期)2020-06-15 11:54:32
故事大王(2019年4期)2019-05-14 16:38:48
城市轨道交通(2019年2期)2019-04-04 08:55:54
中成药(2018年6期)2018-07-11 03:01:32
中西医结合心血管病电子杂志(2017年6期)2017-09-07 21:11:19
中国实用医药(2016年28期)2016-12-07 07:55:18
中国粮油学报(2016年5期)2016-01-23 02:45:06
