
三种核酸碱基化合物密度泛函理论研究
2016-02-08 03:15赵威
化工设计通讯 2016年11期
赵 威
(营口理工学院化学与材料工程系,辽宁营口 115004)
三种核酸碱基化合物密度泛函理论研究
赵 威
(营口理工学院化学与材料工程系,辽宁营口 115004)
运用量子化学密度泛函理论计算了3种核酸碱基化合物胞嘧啶、胸腺嘧啶和尿嘧啶的几何构型、电子结构,运用含时密度泛函计算了3种化合物的电子光谱性质。研究结果表明:胸腺嘧啶化合物结构最稳定,胞嘧啶最大吸收波长红移最明显。
密度泛函;核酸碱基;胞嘧啶;红移
1 引言
核酸碱基主要是指嘌呤和嘧啶的衍生物,是核酸、核苷、核苷酸的成分。构成核酸的碱基有胞嘧啶、胸腺嘧啶、鸟嘧啶、腺嘌呤和鸟嘌呤,胞嘧啶,学名为2-羰基-4-氨基嘧啶,分子式为C4H5N3O。核酸(DNA和RNA)中的主要碱基组成成分之一。胸腺嘧啶是DNA的主要嘧啶碱,在RNA中极少见;相反,尿嘧啶是RNA的主要嘧啶碱,在DNA中则是稀有的。目前对核酸碱基化合物的设计研究的文献很多,研究表明天然碱基化合物基本都不具有荧光活性,但是可以对这类化合物用碱基修饰来寻找具有高荧光活性的化合物,比如引入芳香环等[1]。本文运用量子化学密度泛函理论计算了3种核酸碱基化合物胞嘧啶、胸腺嘧啶和尿嘧啶的几何构型、电子结构,运用含时密度泛函计算了三种化合物的电子光谱性质。3种核酸碱基化合物的结构图见图1。
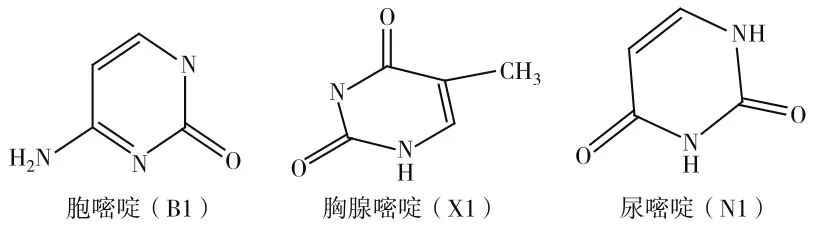
图1 三种核酸碱基化合物的结构图
2 计算方法
运用Guassian-03W量子化学程序包中的密度泛函(DFT)理论[2],在B3LYP/6-31G*水平上,对(图1)进行全自由度几何优化,频率计算。然后对三种化合物的基态优化构型为基础,应用含时密度泛函理论TD-DFT[3],在B3LYP/6-31G(d,p)水平上,计算了三种化合物在气相中的电子吸收光谱。所有计算均采用Guassian-03W程序完成。
3 计算结构与讨论
1)三种核酸碱基化合物能量计算结果,三种化合物的结构优化得到的能量数据见表1。
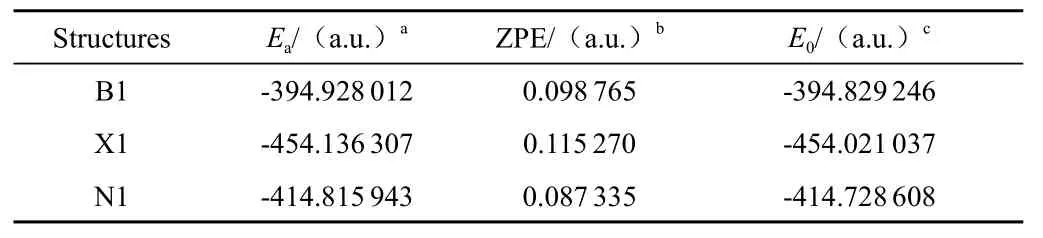
表1 三种核酸碱基化合物能量
Ea:Total electronic energy;ZPEb:Zero-point energy;E0:Sum of electronic and zero-point energyE0是经零点能校正后的总能量,无特别说明均以E0为能量讨论的依据。从表1数据显示胸腺嘧啶能量最低-454.021 037a.u,胞嘧啶的能量最高为-394.829 246a.u,尿嘧啶的能量-414.728 608a.u,介于两者之间。
(2)三种核酸碱基化合物的振动频率和强度。三种化合物的结构优化得到的振动频率和振荡强度数据见表2。

表2 三种核酸碱基化合物振动频率和振荡强度
频率计算结果表明,胞嘧啶的振动频率138.136cm-1,振荡强度1.601 3km/mol,胸腺嘧啶的振动频率119.959cm-1,振荡强度0.001 1km/mol,尿嘧啶的振动频率148.456cm-1,振荡强度0.368 8km/mol,三种化合物的振动频率,最小频率都为正值,没有出现虚频,进一步证明这三种化合物稳定的存在。
(3)三种核酸碱基化合物化合物的跃迁轨道、成分、最大吸收波长(λ)、振子强度(f)和主要跃迁类型见表3。

表3 三种核酸碱基化合物的跃迁轨道、成分、最大吸收波长(λ)、振子强度(f)和主要跃迁类型
表3数据显示胞嘧啶的跃迁类型是π-π*,主要是对应HOMO-LUMO的跃迁,跃迁成分是65.31%,跃迁对应的最大吸收波长是266.23nm,对应的振子强度是0.044,胸腺嘧啶的跃迁类型是π-π*,主要是对应HOMO-1-LUMO的跃迁,跃迁成分是67.34%,跃迁对应的最大吸收波长是260.79nm,对应的振子强度是0.001,尿嘧啶的跃迁类型是π-π*,主要是对应HOMO-1-LUMO的跃迁,跃迁成分是67.70%,跃迁对应的最大吸收波长是263.40nm,对应的振子强度是0.001。3种核酸碱基化合物的的最大吸收波长均在紫外波范围内。
[1] 张来斌、任廷琦.新型鸟嘌呤类似物y-鸟嘌呤及其异构体电子光谱性质的理论研究[J].物理学报,2015,64(7).
[2] Kohn W,Sham L.Self-consistent equations including exchange and correlation effects[J].Phys Rev A,1965,140:1133.
[3] Petersila M,Gossmann U J,Gross E K U.Excitation energies from time-dependent density-functional theory[J].Phys Rev Lett,1996,76:1212.
Density Functional Theory Study on Three Kinds of Nucleic Acid base Compounds
Zhao Wei
In this paper,by using quantum chemical DFT theory of three kinds of nucleic acid base compound cytosine,thymine and uracil geometry,electronic structure and electronic spectrum properties of three compounds were calculated by using time-dependent density functional theory,the results of the study show that the most stable structure of complexes of thymine,cytosine bathochromic shift in the maximum absorption wavelength is most obvious.
DFT;Nucleic acid;cytosine;red-shift
0643
A
1003-6490(2016)11-0061-01
2016-10-26
赵威(1983—),女,辽宁盖州人,硕士研究生,实验师,主要从事物理化学实验教学以及理论化学研究。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
农药科学与管理(2019年6期)2019-11-23
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
武警医学(2018年10期)2018-11-06
西安文理学院学报(自然科学版)(2016年4期)2016-12-19
百科知识(2015年13期)2015-09-10
