
C-H活化策略高效合成新型吲哚[2,1-a]异喹啉化合物
2016-01-08 08:17王亮,陈玉婷,杜依娜等
化学与生物工程 2015年9期
关键词:吲哚
C-H活化策略高效合成新型吲哚[2,1-a]异喹啉化合物
王亮,陈玉婷,杜依娜,顾昌翰,彭望明
(江汉大学 光电化学材料与器件省部共建教育部重点实验室,湖北 武汉430056)
摘要:以吲哚为原料,使用容易安置和移除的嘧啶基作为导向基团,以C-H活化/芳化反应为关键步骤,实现了中间体N-嘧啶-2-苯基吲哚的高效合成。通过简便的方法移除该中间体上的嘧啶导向基团,以C-H活化/环化反应为关键步骤,实现了新型吲哚[2,1-a]异喹啉的构建。对C-H活化/芳化反应条件进行了优化,并在优化的反应条件下进行了该反应的放大量实验。所有化合物均采用IR、NMR、HRMS等多种谱学技术进行了结构表征。
关键词:吲哚;导向基团;C-H活化;多环吲哚
基金项目:国家自然科学基金资助项目(21302064),湖北省教育厅科学研究计划指导性项目(B2015231),江汉大学博士科研启动经费资助项目(2012023)
收稿日期:2015-05-25
作者简介:王亮(1982-),男,湖北人,博士研究生,研究方向:过渡金属催化的C-H活化反应,E-mail:wangliang@jhun.edu.cn。
doi:10.3969/j.issn.1672-5425.2015.09.005
中图分类号:TQ 251.34文献标识码:A
多环吲哚及其衍生物是一类重要的含氮杂环化合物,是来源于自然界的天然生物碱,广泛分布于动植物和生物真菌界中。这类化合物大多数具有显著的生理和药理活性,已被应用于各种医药制剂、染料和精细化学品等的研发与利用[1-2]。据不完全统计,含有多环吲哚结构单元的天然产物有3 000多种,其中40多种已应用于临床医疗[3]。因此,具有多环吲哚骨架的化合物被认为是发展新型药物先导分子的“优势结构”,其化学合成一直吸引着化学家们的广泛研究兴趣[4]。
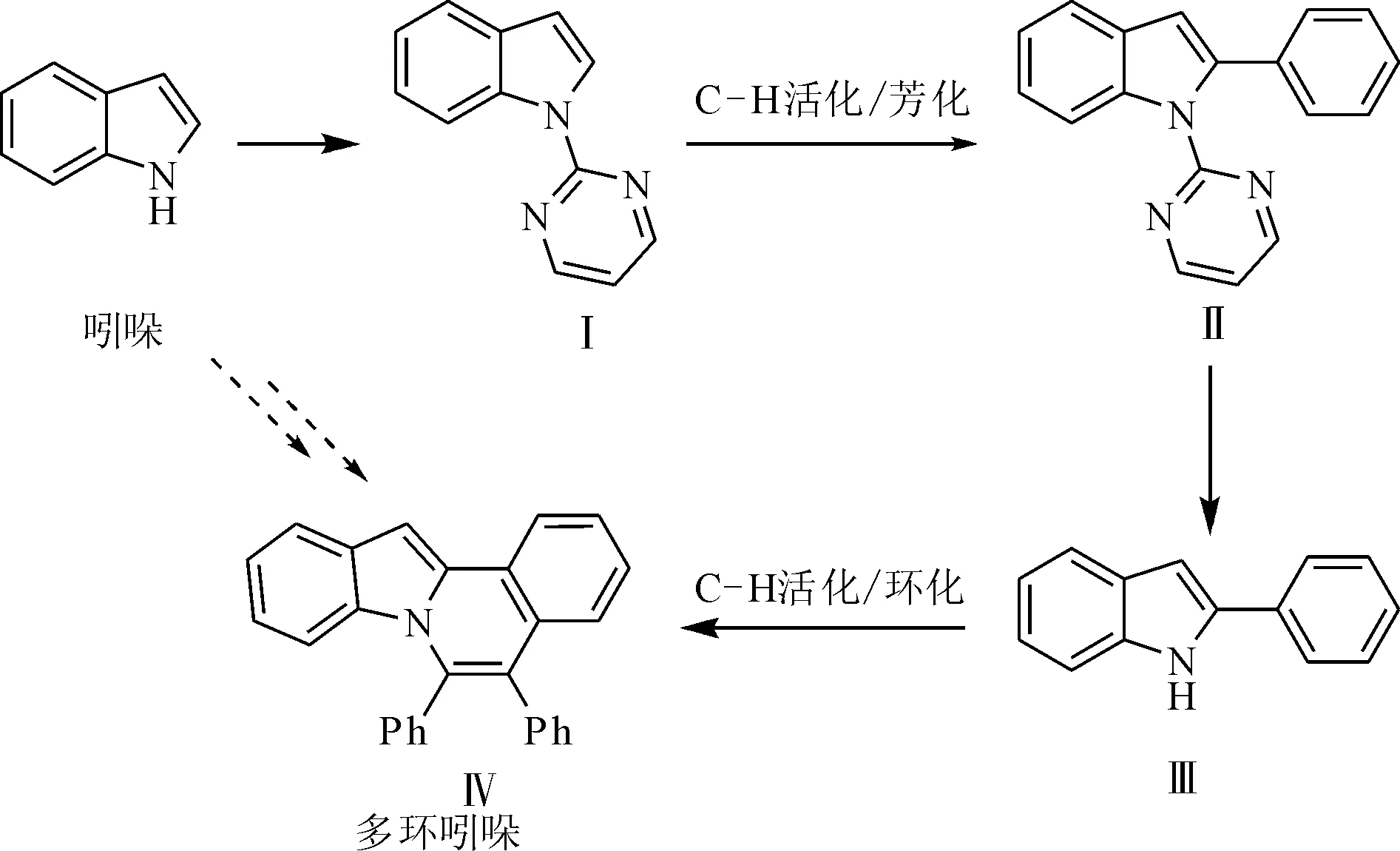
图1 吲哚[2,1- a]异喹啉的合成路线 Fig.1 Synthetic route of indolo[2,1- a]isoquinoline
1实验
1.1 试剂与仪器
吲哚、Cu(OAc)2·H2O,天津希恩思生化科技有限公司;氯嘧啶,上海柏卡化学技术有限公司;无水碳酸钠、无水硫酸镁、氯化钠、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、二甲苯、甲醇、乙醇、石油醚、乙酸乙酯,天津福晨化学试剂厂;三氟乙酸银、苯硼酸,上海阿拉丁化学试剂有限公司;[RhCp*Cl2]2,Alfa-Aesar试剂有限公司;乙醇钠,上海迈瑞尔化学技术有限公司;二苯基乙炔、氢化钠(60%),萨恩化学技术(上海)有限公司。所用试剂为分析纯或化学纯,在未作任何特殊声明的情况下,所用试剂和溶剂都经过标准方法纯化后使用。
Bruker Avance Ⅱ-400 MHz型超导核磁共振仪、Bruker microTOF-QⅡ型高分辨质谱仪、Bruker TENSOR27 型傅立叶变换红外光谱仪(直接涂片测定),德国布鲁克(Bruker)公司;WRS-2 型微机熔点仪,上海圣科仪器设备有限公司;ZF-20D型暗箱式紫外分析仪、DF-101S型集热式磁力搅拌器,巩义予华仪器有限公司; BSA224S型电子天平,赛多利斯(上海)贸易有限公司;RE2000A型旋转蒸发仪,上海亚荣生化仪器厂。
1.2 方法
1.2.1化合物Ⅰ(N-嘧啶吲哚)的合成
N2保护下,在50 mL两口瓶内加入吲哚(5.0 mmol,585.7 mg),并溶于DMF(15 mL)中,搅拌均匀后冰浴冷却。向溶液中加入NaH(6.0 mmol,240.0 mg),自然升至室温继续搅拌0.5 h。再次冰浴冷却,向体系中缓慢滴加氯嘧啶(6.0 mmol,687.2 mg)的DMF(5 mL)溶液,加毕后置于130 ℃油浴中反应。薄层层析色谱法(TLC)检测至反应完全,向体系中加入H2O(15 mL)淬灭反应,随后转至分液漏斗,用乙酸乙酯(15 mL×2)萃取,合并有机相,用饱和氯化钠溶液(20 mL×1)洗涤。分液后,用无水硫酸镁干燥,减压除去有机溶剂,用硅胶柱层析分离纯化[V(石油醚)∶V(乙酸乙酯)=8∶1],即得到纯净的产物,收率 85%。
1.2.2化合物Ⅱ(N-嘧啶-2-苯基吲哚)的合成(C-H活化/芳化反应)
在10 mL的Schlenk反应管中依次加入化合物Ⅰ(0.2 mmol,39.0 mg)、苯硼酸(0.4 mmol,48.8 mg)、三氟乙酸银(0.8 mmol,176.7 mg)、催化剂[RhCp*Cl2]2[1%(摩尔分数),1.24 mg]以及CH3OH(1.0 mL)。置于60 ℃油浴中反应,TLC检测反应至完全。反应混合物用硅藻土滤去不溶物,用CH2Cl2(15 mL)洗涤,减压除去有机溶剂,用硅胶柱层析分离纯化 [V(石油醚)∶V(乙酸乙酯)= 5∶1],即得到纯净的产物,收率98%。
1.2.3化合物Ⅲ(2-苯基吲哚)的合成
N2保护下,在50 mL两口瓶内加入化合物Ⅱ(2.0 mmol,542.6 mg),并溶于DMSO(15 mL)中,搅拌均匀后冰浴冷却,向溶液中加入EtONa(10.0 mmol,680.0 mg)。加毕后置于110 ℃油浴中反应,TLC检测反应至完全。向体系中加入H2O(15 mL)淬灭反应,随后转至分液漏斗,用乙酸乙酯(15 mL×3)萃取,合并有机相,用饱和氯化钠溶液(20 mL×1)洗涤。分液后,用无水硫酸镁干燥,减压除去有机溶剂,用硅胶柱层析分离纯化 [V(石油醚)∶V(乙酸乙酯)=3∶1],即得到纯净的产物,收率 99%。
1.2.4化合物Ⅳ的合成(C-H活化/环化反应)
在10 mL的Schlenk反应管中依次加入化合物Ⅲ(0.25 mmol,48.3 mg)、二苯基乙炔(0.30 mmol,53.5 mg)、催化剂[RhCp*Cl2]2(0.005 mmol,3.0 mg)、Cu(OAc)2·H2O (0.025 mmol,5.0 mg)、 Na2CO3(0.50 mmol,53.0 mg)以及二甲苯(1.5 mL),密封后安置O2气球(800 mL)并置于100 ℃油浴中反应,TLC检测反应至完全。反应混合物用硅藻土滤去不溶物,用CH2Cl2(20 mL)洗涤,减压除去有机溶剂,用硅胶柱层析分离纯化 [V(石油醚)∶V(乙酸乙酯)=2∶1],即得到纯净的产物,收率 95%。
2结果与讨论
2.1 化合物的结构表征
化合物Ⅰ,白色固体,收率85%,m.p.85~86 ℃。 IR,ν,cm-1:1 570,1 535,1 465,1 312,1 089,1 064,982,915,773,741,722;1HNMR(400 MHz,CDCl3),δ: 8.85 (d,J=8.4 Hz,1H),8.64 (d,J=4.8 Hz,2H),8.26 (d,J=3.7 Hz,1H),7.64 (d,J=7.7 Hz,1H),7.33 (t,J=7.7 Hz,1H),7.25 (t,J=7.5 Hz,1H),7.07 (t,J=4.8 Hz,1H),6.70 (d,J=3.6 Hz,1H);13CNMR (100 MHz,CDCl3),δ: 158.1,157.8,135.4,131.3,125.8,123.7,122.0,120.9,116.3,116.2,106.9; HRMS,m/z: calcd for C12H9N3Na [M+Na+] 218.0689,found 218.0693。
化合物Ⅱ,白色固体,收率98%,m.p.126~127 ℃。 IR,ν,cm-1:1 560,1 428,1 365,898,845,818,763,742,690;1HNMR (400 MHz,CDCl3),δ: 8.66 (d,J=4.8 Hz,2H),8.13 (d,J=8.1 Hz,1H),7.65 (d,J=7.7 Hz,1H),7.35~7.23 (m,7H),7.10 (t,J=4.8 Hz,1H),6.81 (s,1H);13CNMR (400 MHz,CDCl3),δ: 158.2,158.2,140.5,138.1,134.0,129.4,128.2,128.1,127.2,123.6,122.2,120.7,117.6,112.8,108.2; HRMS,m/z:calcd for C18H14N3[M+H+] 272.1182,found 272.1187。
化合物Ⅲ,白色固体,收率99%,m.p.178~179 ℃。IR,ν,cm-1:3 445,1 565,1 457,873,748,689;1HNMR (400 MHz,CDCl3),δ:8.29 (s,1H),7.64 (t,J=7.4 Hz,3H),7.50~7.36 (m,3H),7.31 (t,J=7.4 Hz,1H),7.20 (dd,J=15.7,8.5 Hz,1H),7.12 (t,J=7.4 Hz,1H),6.82 (d,J=1.2 Hz,1H);13CNMR (400 MHz,CDCl3),δ:137.9,136.8,132.4,129.3,129.0,127.8,125.1,122.2,120.7,120.3,111.0,99.9;HRMS,m/z:calcd for C18H14N3[M+H+] 272.1182,found 272.1187。
化合物Ⅳ,淡黄色固体,收率95%,m.p.224~225 ℃。IR,ν,cm-1:1 573,1 445,1 380,1 233,1 067,898,712,684;1HNMR (400 MHz,CDCl3),δ:8.30 (d,J=8.0 Hz,1H),7.79 (d,J=8.0 Hz,1H),7.50 (t,J=8.6 Hz,1H),7.41 (s,1H),7.39~7.29 (m,6H),7.27~7.13 (m,7H),6.81 (t,J=7.8 Hz,1H),5.99 (d,J=8.7 Hz,1H);13CNMR (400 MHz,CDCl3),δ:136.7,136.0,135.8,135.2,132.6,131.7,130.7,130.2,129.6,128.6,128.5,127.7,127.2,127.0,126.6,126.2,125.3,123.2,121.5,121.3,120.2,114.5,94.1;HRMSm/z:calcd for C28H19N [M+H+] 370.1590,found 370.1598。
2.2 C-H活化/芳化反应条件的优化
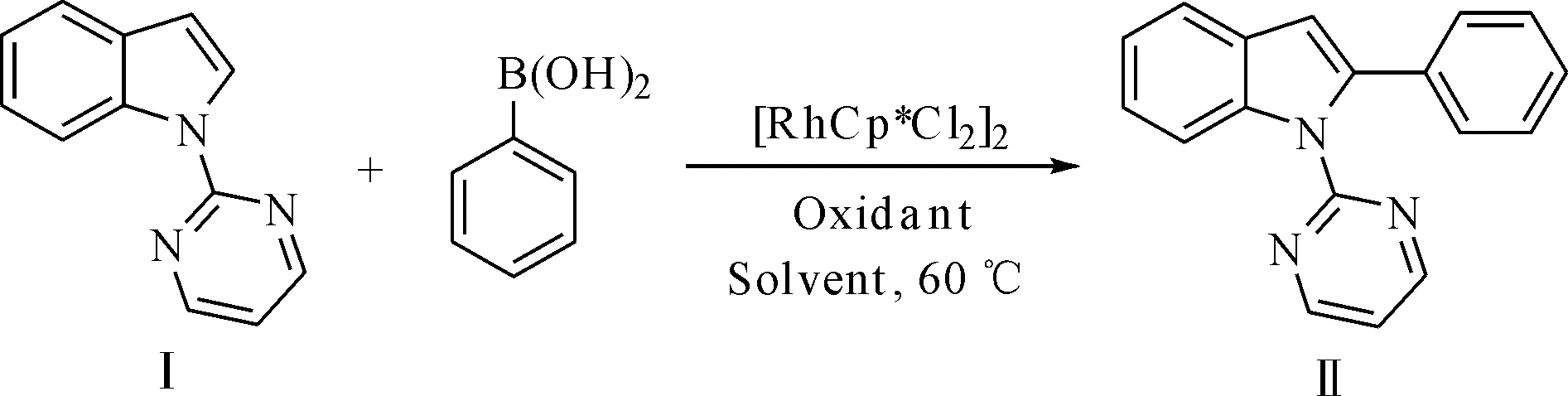
2.2.1氧化剂对反应的影响
在获得了化合物Ⅰ的基础上,对反应关键步骤C-H活化/芳化反应进行简单的条件优化。在甲醇作为溶剂、60 ℃的反应条件下,使用过渡金属[RhCp*Cl2]2作为催化剂,用量为1%(摩尔分数);苯硼酸作为芳基化试剂,用量为2.0 eq.,考察氧化剂对反应的影响,结果见表1。
从表1可以发现,当使用Cu(OAc)2和Ag2O作为氧化剂时,反应24 h后原料未反应完全,收率分别为53%和55%(1#、2#)。当使用AgOAc作为氧化剂时,反应能够在24 h内完成,收率提高到91%(3#)。当使用AgNO3和Ag2CO3作为氧化剂时,反应收率不太理想(4#、5#)。当氧化剂更换为AgOOCCF3时,反应能够在6 h内完成,并且收率进一步提高到98%(6#)。随后,对AgOOCCF3用量进行了考察。当AgOOCCF3用量降低为3.5 eq.时,反应过程中有少量原料未转化完全,收率降低为90%,且反应时间延长到12 h(7#);当继续降低AgOOCCF3用量时,原料不仅没有转化完全,并且反应时间也很长,收率显著下降(8#、9#)。可见使用银金属氧化剂的阴离子以及氧化剂的用量对反应具有较大的影响。
表1氧化剂对C-H活化/芳化反应的影响
Tab.1Effect of oxidants on the C-H activation/arylation

实验号氧化剂用量/eq.反应时间/h收率/%1#Cu(OAc)24.024532#Ag2O4.024553#AgOAc4.024914#AgNO34.012255#Ag2CO34.024596#AgOOCCF34.06987#AgOOCCF33.512908#AgOOCCF33.024789#AgOOCCF32.02451
注:反应条件:化合物Ⅰ用量0.2 mmol,苯硼酸用量0.4 mmol,[RhCp*Cl2]21.0%(摩尔分数),甲醇1.0 mL,反应温度60 ℃;收率为分离收率。
2.2.2溶剂对反应的影响(表2)
表2溶剂对C-H活化/芳化反应的影响
Tab.2Effect of solvents on the C-H activation/arylation
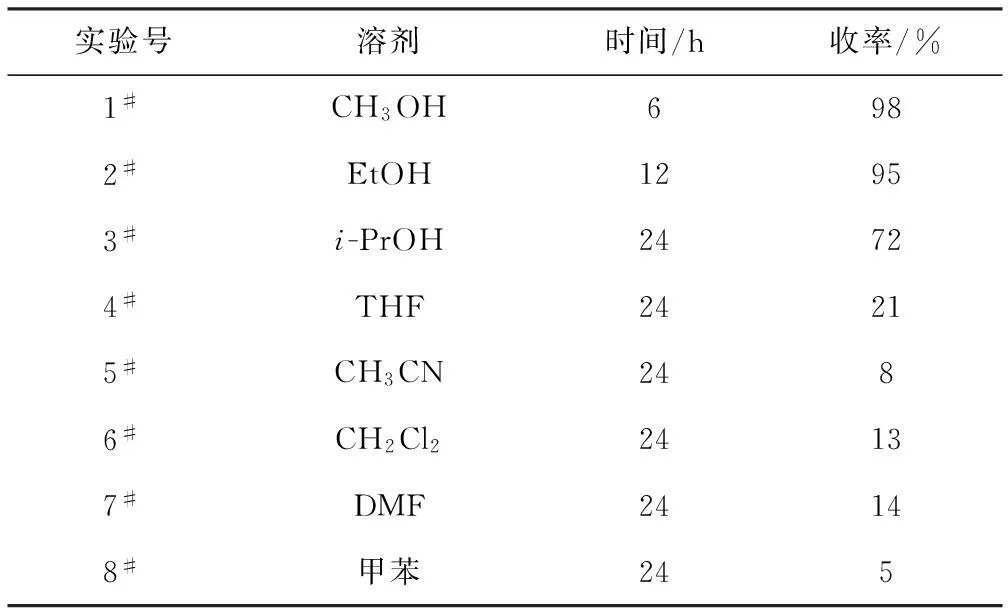
实验号溶剂时间/h收率/%1#CH3OH6982#EtOH12953#i-PrOH24724#THF24215#CH3CN2486#CH2Cl224137#DMF24148#甲苯245
注:反应条件:化合物Ⅰ用量0.2 mmol,苯硼酸用量0.4 mmol,[RhCp*Cl2]21.0%(摩尔分数),AgOOCCF3用量0.8 mmol,溶剂1.0 mL,反应温度60 ℃;收率为分离收率。
从表2可以发现,当使用甲醇为溶剂时,反应可在6 h内完成,收率高达98%(1#)。当改用乙醇为溶剂时,反应在12 h内完成,收率为95%(2#)。当改用异丙醇为溶剂时,收率明显下降,为72%(3#)。THF、CH3CN、CH2Cl2、DMF以及甲苯都不是很好的溶剂,反应收率均较低(4#~8#)。可见溶剂对反应的影响较大,在质子性溶剂中反应能获得较好的效果。
2.2.3放大量实验
将反应底物化合物Ⅰ的用量由0.2 mmol扩大到5.5 mmol,达到克级规模进行放大量实验(图2)。反应仍能够顺利进行且反应效率没有受到影响,以96%的收率获得C-H活化/芳化的产物。因此,铑催化的C-H活化/芳化反应在某些程度能够进行放大量实验,为多环吲哚的大规模生产提供了一定实验基础。
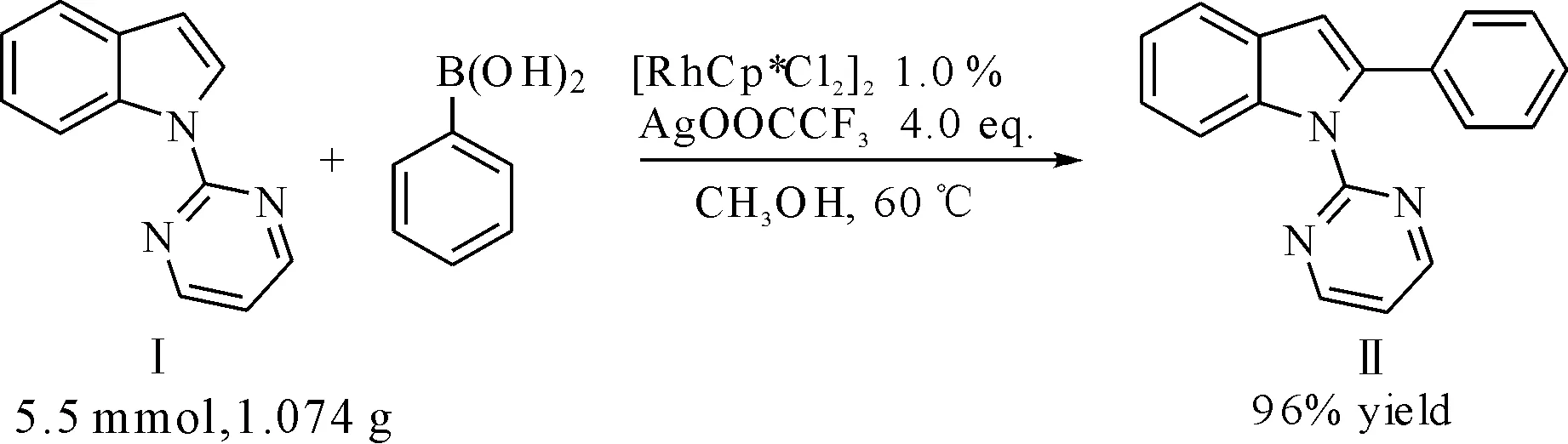
图2 克级规模的实验 Fig.2 Gram-scale experiment
3结论
通过简单易得的吲哚为起始原料,使用容易安置和移除的嘧啶作为导向基团,以C-H活化/芳化反应为关键步骤,实现了N-嘧啶-2-苯基吲哚的合成。通过简便方法移除该中间体上的嘧啶导向基团,以C-H活化/环化反应为关键步骤,实现了新型吲哚[2,1-a]异喹啉的高效构建。所有化合物均采用IR、NMR、HRMS进行了结构表征。
参考文献:
[1]SOMEI M,YAMADA F.Simple indole alkaloids and those with a non-rearranged monoterpenoid unit[J].Natural Product Reports,2005,22(1):73-103.
[2]HUMPHREY G R,KUETHE J T.Practical methodologies for the synthesis of indoles[J].Chemical Reviews,2006,106(4):2875-2911.
[3]BANDINI M,EICHHOLZER A.Catalytic functionalization of indoles in a new dimension[J].Angewandte Chemie International Edition,2009,48(5):9608-9644.
[4]张红.吲哚衍生物合成方法研究新进展[J].化学与粘合,2010,32(3):42-45.
[5]DAVIES H M L,MANNING J R.Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion[J].Nature,2008,451(7177):417-424.
[6]LEOW D,LI G,MEI T S,et al.Activation of remote meta-C-H bonds assisted by an end-on template[J].Nature,2012,486(7404):518-522.
[7]LYONS T W,SANFORD M S.Palladium-catalyzed ligand-directed C-H functionalization reactions[J].Chemical Reviews,2010,110(2):1147-1169.
[8]ACKERMANN L.Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations:Mechanism and scope[J].Chemical Reviews,2011,111(3):1315-1345.
[9]MCMURRAY L,O′HARA F,GAUNT M J.Recent developments in natural product synthesis using metal-catalysed C-H bond functionalisation[J].Chemical Society Reviews,2011,40(12):1885-1898.
[10]DING S,SHI Z,JIAO N.Pd(Ⅱ)-catalyzed synthesis of carbolines by iminoannulation of internal alkynesviadirect C-H bond cleavage using dioxygen as oxidant[J].Organic Letters,2010,12 (7):1540-1543.
[11]WANG L,GUO W,ZHANG X X,et al.Synthesis of indolo [1,2-a]quinoxalinesviaa Pd-catalyzed regioselective C-H olefination/cyclization sequence[J].Organic Letters,2012,14(3):740-743.
Highly Efficient Synthesis of A Novel Indolo[2,1-a]isoquinoline
DerivativeviaC-H Activation Strategy
WANG Liang,CHEN Yu-ting,DU Yi-na,GU Chang-han,PENG Wang-ming
(KeyLaboratoryofOptoelectronicChemicalMaterialsandDevicesofMinistryof
Education,JianghanUniversity,Wuhan430056,China)
Abstract:The intermediate N-pyrimidyl-2-phenyl-indole was highly efficient synthesized with indole as raw material and the easily installable and removable pyrimidyl group as directing group.The C-H activation/arylation was the key step for the synthesis of N-pyrimidyl-2-phenyl-indole compound.After removal of the pyrimidyl group under simple reaction conditions,the novel indolo[2,1-a]isoquinoline derivative could be successfully synthesized via C-H activation/cyclization.The optimal reaction conditions were established by examining the effect of oxidants and solvents.The gram-scale experiments was conducted under the optimal reaction conditions.The structures of all compounds were characterized by IR,NMR and HRMS.
Keywords:indole;directing group;C-H activation;polycyclic indoles
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
生物技术通报(2021年12期)2021-02-10
昆明医科大学学报(2020年12期)2021-01-26
太原师范学院学报(自然科学版)(2020年3期)2020-08-13
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
山东化工(2019年11期)2019-06-26
国际呼吸杂志(2019年1期)2019-03-08
合成化学(2017年9期)2017-09-16
中国药理学与毒理学杂志(2015年3期)2015-12-16
