
W-Fe磷桥联配合物CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3的结构和性质
2015-12-28 07:44史小琴李亚平
徐州工程学院学报(自然科学版) 2015年3期
关键词:金属
王 菊,史小琴,李亚平
(1.徐州工程学院,江苏 徐州 221018;2.北京化工大学 理学院,北京 100029)
W-Fe磷桥联配合物CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3的结构和性质
王菊1,史小琴1,李亚平2
(1.徐州工程学院,江苏 徐州221018;2.北京化工大学 理学院,北京100029)
摘要:文章采用B3LYP、B3P86、MPW1PW91、M06等密度泛函方法对CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3进行几何结构全优化及频率分析以确定其平衡构型的热力学稳定性.通过比较发现,密度泛函方法M06能够更好地评价W-Fe双金属磷桥联配合物.CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在较弱的金属-金属(W-Fe)共价单键,成键方式主要为BD (1)W-Fe→BD*(1)W-Fe.金属W及Fe分别与羰基成键,金属W与羰基间存在较强的共价单键,而金属Fe与羰基键较弱,是弯曲的,其键曲率大于1.
关键词:密度泛函理论;金属-金属键;金属-羰基键;自然键轨道理论;分子电荷密度拓扑理论
双核金属配合物的突出特点是金属-金属键(给体-受体键)在化学反应中起到“开关”控制作用,即:金属-金属键断裂提供空位使配体连接到配合物上,金属-金属键重新形成提供了进一步反应的驱动力,这种现象被称为相邻双金属的协同作用[1-2].根据这一性质,在合成双核金属配合物时,可以通过精心选择不同种类金属和配体来控制化学反应位置,因此,双核金属配合物被广泛地应用于石油、催化、汽车等行业[3-5].
近年来,越来越多的研究工作利用自然键轨道理论[6]和分子电荷密度拓扑理论[7-8]对分子结构、性质及反应机理进行研究[9-11].本文采用4种密度泛函方法对CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3进行几何结构全优化及频率分析以确定其平衡构型的热力学稳定性.通过与实验测定数据进行比较,确定几种密度泛函方法在评价W-Fe磷桥联配合物上的优劣.采用自然键轨道理论及分子电荷密度拓扑理论研究双金属磷桥联配合物CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中金属-金属(W-Fe)键及金属-羰基(W-C=O及Fe-C=O)性质.
1计算方法
采用B3LYP[12]、B3P86[13]、MPW1PW91[14]、M06[15]等密度泛函方法对CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3进行几何结构全优化及频率分析以确定其平衡构型的热力学稳定性,其中:W、Fe等原子选用LanL2Dz基组,其余原子P、C、O、H等选用6-311++G(d,p)基组.将4种方法优化得到的配合物几何结构与实验测定数据进行比较发现,密度泛函方法M06更适合于计算CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3体系,能够提供与实验测定数据更为接近的计算结果.在M06几何结构全优化所得配合物CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3平衡构型基础上,采用自然键轨道理论[6]和分子电荷密度拓扑理论[7-8]研究W-Fe键及金属-羰基(W-C=O及Fe-C=O)的性质.
全部计算工作在单台计算机上采用Gaussian09软件包[16]完成,分子电荷密度拓扑分析采用AIM2000软件[17]完成.
2结果与讨论
2.1 平衡构型
表1给出B3LYP、MPW1PW91、M06、B3P86等密度泛函方法进行几何结构全优化得到CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3平衡构型的部分键长、键角数据.为确定上述方法在评价W-Fe磷桥联配合物上的优劣,将4种密度泛函方法优化得到的几何结构计算值与CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3相关的实验测定值[1]进行比较,计算值与理论值的差异见表1.通过比较发现,键长平均误差依次为:B3LYP(3.07%)、MPW1PW91(2.69%)、M06(1.81%)、B3P86(2.21%),键角平均误差依次为:B3LYP(2.38%)、MPW1PW91(2.28%)、M06(1.21%)、B3P86(2.33%),其中,密度泛函方法M06优化得到的CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3计算值更接近于实验测定值.

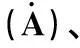
表1 CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3几何结构数据a(包括部分键长键角(o)

续表1

图1 CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3平衡构型(M06/GEN)
2.2 金属-金属(W-Fe)键性质
表2给出CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3部分键临界点的性质.在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中,金属(W)与金属(Fe)之间存在(3,-1)键临界点[7-8],即:(3,-1)BCPW-Fe.(3,-1)BCPW-Fe键临界点性质如下:电子密度为(小于正常的C—C键的电子密度为,拉普拉斯为,键曲率为0.709 38,Hb为-0.008 44 a.u.(Hb<0),-Gb/Vb为0.711 45(0.5<-Gb/Vb<1.0).上述AIM分析结果表明,在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在较弱的共价金属-金属(W-Fe)键.W-Fe键曲率(0.709 387)小于1,说明金属W与金属Fe间不存在π电子的相互作用,金属-金属(W-Fe)键为单键.
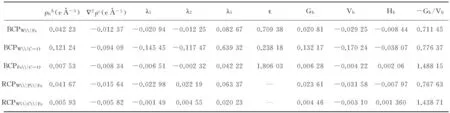
表2 CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3部分键临界点的性质
此外,对CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3进行了自然键轨道分析,表3给出W-Fe成(反)键轨道原子轨道杂化贡献.从表3可以发现,W原子参与杂化的原子轨道主要包括6p轨道(占24.86%)和5d轨道(占69.77%),Fe原子参与杂化的原子轨道主要是4p轨道(占45.85%)和3d轨道(占40.77%).

表3 金属-金属(W-Fe)成(反)键轨道原子轨道杂化贡献
表4给出金属-金属(W-Fe)键的自然键轨道分析结果,从表4可以看出,成键轨道BD(1)W-Fe与BD*(1)W-Fe、RY*(1) P3、RY*(1) W1、RY*(1)Fe存在相互作用,其中,相互作用模式BD (1)Mo-Fe→BD*(1)Mo - Fe二阶微扰稳定能[6]最大,为15.13 kcal/mol,表明金属-金属(W-Fe)键的成键方式主要为BD(1)W-Fe→BD*(1)W-Fe.
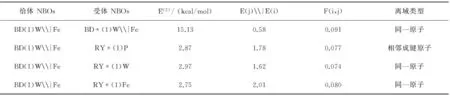
表4 金属-金属(W-Fe)键自然键轨道分析结果
AIM及NBO结果均表明,双金属磷桥联配合物CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在较弱的金属-金属(W-Fe)共价单键,该金属-金属(W-Fe)键的成键方式主要为BD (1)W-Fe→BD*(1)W-Fe.
2.3 金属-羰基(W-C=O及Fe-C=O)性质
从表2可以看出,CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中,金属Fe及W分别与羰基碳原子间存在一个(3,-1)键临界点,这说明金属Fe及金属W分别与羰基(C6=O6)成键.
W-C6=O6键临界点性质:电子密度为(略小于正常的C-C单键的电子密度,拉普拉斯为,键曲率为0.238 18,Hb为-0.03807 a.u.(Hb<0),-Gb/Vb为0.776 37(0.5<-Gb/Vb<1.0).Fe-C6=O6键临界点性质:电子密度为(远小于正常的C-C单键的电子密度,拉普拉斯为,键曲率为1.806 03,Hb为0.002 06a.u.(Hb>0),-Gb/Vb为1.488 15.
AIM分析结果表明,CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在较强的共价W-C6=O6键,W-C6=O6键曲率(0.238 18)小于1,说明金属W与羰基C=O间不存在π电子的相互作用,W-C=O键为单键.而Fe-C6=O6键较弱,键曲率大于1,说明该键是弯曲的,并且金属Fe与羰基间存在π电子的相互作用.以上分析表明,尽管金属W与Fe都能与羰基成键,但是两者的性质是截然不同的.
在W-P-Fe及W-羰基(C6=O6)-Fe间分别存在一个(3,+1)环临界点[7-8],这说明在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在环状结构W-P-Fe及W-羰基(C6=O6)-Fe.从表2可以看出,(3,+1)RCPW-P-Fe环临界点的性质:电子密度为,拉普拉斯为为-0.007 97 a.u.(Hb<0),-Gb/Vb为0.767 63(0<-Gb/Vb<1.0);(3,+1)RCPW-C-Fe环临界点的性质:电子密度为(远小于环状结构W-P-Fe环临界点电子密度),拉普拉斯为为-0.003 10 a.u.(Hb<0),-Gb/Vb为1.438 71(-Gb/Vb>1.0).通过比较发现,尽管在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在W-P-Fe及W-羰基(C=O)-Fe等两种环状结构,但W-P-Fe环状结构较W-羰基-Fe环状结构在热力学上更为稳定.
3结论
采用B3LYP、B3P86、MPW1PW91、M06等密度泛函方法对CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3进行几何结构全优化及频率分析以确定其平衡构型的热力学稳定性.通过比较发现,M06优化得到的CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3几何结构计算值更接近于实验测定值.AIM及NBO结果表明,在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在较弱的金属-金属(W-Fe)共价单键,该金属-金属(W-Fe)键的成键方式主要为BD(1)W-Fe→BD*(1)W-Fe.金属W及Fe能够分别与羰基成键,但两者的性质是截然不同的.金属W与羰基间存在较强的共价单键,而金属Fe与羰基成键较弱,键曲率大于1(说明该键是弯曲的,金属Fe与羰基间存在π电子的相互作用).此外,在CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3中存在环状结构W-P-Fe及W-羰基(C6=O6)-Fe,W-P-Fe环状结构较W-羰基(C6=O6)-Fe环状结构在热力学上更为稳定.
参考文献:
[1] Shyu S G,Hsiao S M,Lin K J,et al.Synthesis,structure,fluxional behavior,and substitution reaction of the metal-metal-bonded heterobimetallic phosphido-bridged complex[J].Organometallics,1995,14(9):4300-4307.
[2] Shyu S G,Lin P J,Wen Y S.Synthesis,structure and chemistry of heterobiometallic phosphido-bridged complexes CpFe(CO)2(μ-PPh2)M(CO)5(M=Cr,Mo,W)[J].J Organometal Chem,1993,443(1):115-121.
[3] Alberto,D B,Nicoleta P.Thermocalalytic hydroconversion of heavy petroleum cuts with dispersed catalyst [J].Applied Catalysis,1993,94(1):1-16.
[4] Hashimoto H,Kurashima K,Tobita H,et al.Reactions of a phosphido-bridged unsymmetrical diiron complex (η 5-C 5 Me 5) Fe 2 (CO) 4 (μ-CO)(μ-PPh 2) with various alkynes[J].Journal of organometallic chemistry,2004,689(9):1481-1495.
[5] Arifin K,Majlan E H,Daud W R W,et al.Bimetallic complexes in artificial photosynthesis for hydrogen production:A review[J].International Journal of Hydrogen Energy,2012,37(4):3066-3087.
[6] Reed A E,Weinstock R B,Weinhold F.Natural population analysis[J].The Journal of Chemical Physics,1985,83(2):735-746.
[7] Bader R F W.Atoms in molecules:a quantum theory[M].Oxford:Oxford University Press,1990.
[8] 曹维良,张敬畅.分子电荷密度分布的拓扑学性质和分子中原子的定义[J].化学通报,1991(1):30-34.
[9] Cao W L,Marshall T C,Bader R F W.A description of nitrogen-nitrogen bond in terms of charge distributions[J].Chinese Journal of Chemistry,1990,8(6):492-506.
[10] Wang F,Yang H,Yang Z,et al.Geometries and properties of bimetallic phosphido-bridged complex Cp (CO) 2 W (μ-PPh 2) W (CO) 5 and Cp (CO) 3 W (μ-PPh 2) W (CO) 5[J].Chemical Physics,2007,332(1):33-38.
[11] Wang J,Yang Z,Wang X,et al.Theoretical study on conformational conversion of cyclohexane inside two cylindrical molecular capsules[J].Journal of Molecular Structure:THEOCHEM,2006,772(1):39-44.
[12] Becke A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].The Journal of Chemical Physics,1993,98(7):5648-5652.
[13] Perdew J P.Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J].Physical Review B,1986,33(12):8822.
[14] Adamo C,Barone V.Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters:The mPW and mPW1PW models[J].The Journal of Chemical Physics,1998,108(2):664-675.
[15] Zhao Y,Truhlar D G.Comparative DFT study of van der Waals complexes:rare-gas dimers,alkaline-earth dimers,zinc dimer,and zinc-rare-gas dimers[J].The Journal of Physical Chemistry A,2006,110(15):5121-5129.
[16] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian09[CP].Gaussian Inc.,Wallingford CT,2009.
[17] Biegler-KÖnig F,SchÖnbohm J,Derdau R,et al.AIM2000 [CP].Hamilton,Ontario:McMaster University,2002.
(编辑崔思荣)
中图分类号:O641
文献标志码:A
文章编号:1674-358X(2015)03-0047-06
收稿日期:2014-12-03
作者简介:邹翠(1989-),女,湖南衡阳人,硕士,主要从事热动力学和纳米材料研究.
Understanding the Bonding Nature of the Complex
CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3:A Theoretical Study
WANG Ju1,SHI Xiaoqin1,LI Yaping2
(1.School of chemistry and chemical engineering,Xuzhou Institute of Technology,Xuzhou 221018,China;
2.Fauclty of Science,Beijing University of Chemical Technology,Beijing 100029,China)
Abstract:The complex CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3was investigated theoretically by four DFT methods.Computational results revealed that the most accurate structural parameters could be predicted by M06/GEN level of theory. AIM and NBO analysis have been done to probe the W-Fe bond and the metal-carbonyl interaction in the complex CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3.There is a W-Fe bond,a Fe-carbonyl bond and a W-carbonyl bond in the complex,respectively.The formation of the W-Fe bond accompanies the dominant charge transfer interactions BD (1)W-Fe→BD*(1)W-Fe.Compared with the W-carbonyl bond covalent bond,there is a weak bend Fe-carbonyl bond in the complex CpW(CO)2(μ-CO)(μ-PPh2)Fe(CO)3.
Key words:DFT; Metal-metal bond; Metal-carbonyl bond; NBO; AIM
猜你喜欢
小学科学(学生版)(2021年3期)2021-04-13
小哥白尼(趣味科学)(2020年9期)2021-01-18
中学生数理化·高一版(2020年11期)2020-12-14
矿产综合利用(2020年1期)2020-07-24
中国自行车(2018年8期)2018-09-26
现代装饰(2018年2期)2018-05-22
中国资源综合利用(2017年3期)2018-01-22
中国资源综合利用(2017年2期)2018-01-22
Coco薇(2015年5期)2016-03-29
中国钼业(2015年4期)2015-03-10
