
HPLC测定苏黄止咳胶囊中盐酸麻黄碱含量的不确定度评定
2015-12-28 00:42
中国卫生标准管理 2015年28期
HPLC测定苏黄止咳胶囊中盐酸麻黄碱含量的不确定度评定
丰 杰 成强波 陈 娟 陈国辉
目的HPLC法测定苏黄止咳胶囊中盐酸麻黄碱含量的不确定度评定方法。方法通过建立其数学模型,分析不确定度的来源,评定各不确定度因素的影响,获得苏黄止咳胶囊中盐酸麻黄碱含量测定的扩展不确定度和置信水平。结果HPLC法测定苏黄止咳胶囊中盐酸麻黄碱含量的扩展不确定度为0.038 6 mg/粒,盐酸麻黄碱含量的测定结果为(2.22±0.038 6)mg/粒。结论本方法可用于HPLC法测定苏黄止咳胶囊的含量的不确定分析,使测定结果更加可靠。
苏黄止咳胶囊;盐酸麻黄碱;HPLC;不确定度评定分析
测量是为了确定被测量的量值,测量不确定度是说明被测量估计值的不可确定程度或可信程度的参数,可以通过评定得到,测量结果的可用性很大程度上取决于其不确定度的大小。因此,测量结果必须同时包含赋予被测量的值及与该值相关的测量不确定度。
苏黄止咳胶囊主要用于咳嗽变异性哮喘(Cough Variant Asthma,CVA)和感冒后咳嗽的治疗。通过该药多年生产、质量及临床实践证明,苏黄止咳胶囊中盐酸麻黄碱的含量对临床疗效具有重要作用。本研究依据国家食品药品监督管理局YBZ00172008,测定苏黄止咳胶囊中盐酸麻黄碱的含量,参考《JJF1059.1-2012测量不确定度评定与表示》及其他不确定评定资料[1-18],分析影响苏黄止咳胶囊中盐酸麻黄碱含量测定结果的不确定度因素,对各个不确定因素进行评定,报告测量结果,包含赋予被测量的值及与该值相关的测量不确定度。
1 仪器与试药
1.1 仪器
安捷伦Agilent 1200高效液相色谱仪,含在线真空脱气机器(G-1322A),高压四元泵(G-1311A),标准自动进样器(G-1313A),智能化柱温箱(G-1316A),可变波长检测器(G-1313A);XS205电子天平;KQ-250B超声波清洗仪。
1.2 试剂与样品
乙腈为色谱纯,其余试剂均为分析纯,水是高纯水。对照品:盐酸麻黄碱由中国食品药品检定研究院提供,纯度为100.0%,供含量测定。苏黄止咳胶囊由北京海燕药业有限公司提供。
2 实验方法
2.1 色谱条件
SHIMADZU Shim-pack VP-ODS C18柱色谱柱(4.6×150 mm,5 µm);流动相为乙腈-水-磷酸-三乙胺(1.5:98.3:0.1:0.1);流速为1.5 ml/min,检测波长为205 nm,柱温30℃,进样体积10 µl。理论塔板数以盐酸麻黄碱峰计算,应不低于6 000。
2.2 配制及测定方法
2.2.1 对照品溶液的配制 精密称取盐酸麻黄碱对照品约10 mg,置50 ml容量瓶中,用0.l mol/L盐酸溶液溶解稀释、摇匀、定容至刻度。从中精密量取25 ml,置100 ml容量瓶中,用0.l mol/L盐酸溶液稀释、摇匀、定容至刻度,制备成每1 ml中含盐酸麻黄碱50 μg对照品溶液。
2.2.2 供试品溶液的配制 取本品内容物,研细,精密称定0.3 g,置具塞锥形瓶中,精密加入0.l mol/L盐酸溶液25 ml,密塞,称定重量,超声处理(功率100 W,频率20 kHz)30 min,取出放冷,再称定重量,用0.l mol/L盐酸溶液补足减失的重量,摇匀,用微孔滤膜(0.45 μl)滤过,取续滤液,即得供试品溶液。
2.2.3 测定法 按外标法计算盐酸麻黄碱的含量。
3 不确定度评定
测量不确定度的评定步骤可归纳为:(1)明确被测量及其测量方法;(2)建立数学模型或称测量模型,是测量不确定度评定的依据;(3)分析找出所有影响测量不确定度的因素,评定各测量不确定度分量;(4)合成相对不确定度的评定;(5)扩展不确定度的评定;(6)形成不确定度报告。根据以上计算步骤,对本次实验的测量不确定度进行评定。
3.1 建立数学模型(或测量模型)
苏黄止咳胶囊中盐酸麻黄碱含量的计算公式为:
X=(C对×A样×V样×ω)÷(m样×A对)
其中,x为每粒供试品中盐酸麻黄碱的含量(mg/粒);A样为供试品中盐酸麻黄碱的峰面积;V样为供试品溶液稀释后的体积(ml),w为每粒供试品的平均装量(g/粒);m样为供试品的称样量(g);A对为对照品盐酸麻黄碱的色谱峰面积;C对为对照品盐酸麻黄碱的浓度(mg/ml)。
C对=(m对×V2)÷(V1×V3)
公式中,m对为对照品盐酸麻黄碱的称样量(mg);V1、V2、V3分别为盐酸麻黄碱对照品第一次定容的体积、稀释时移取对照品溶液的体积、最后稀释后的体积(ml)。
3.2 各个不确定度分量的评定
3.2.1 对照品溶液配制和对照品色谱峰引入的不确定度 对照品溶液配制引入的不确定度来源于对照品称量引入的不确定度和稀释过程引入的不确定度,而称量引入的不确定度来源于对照品纯度和对照品的称量。
(1)对照品纯度引入的相对不确定度:由中国食品药品检定研究院提供的盐酸麻黄碱对照品,纯度为100.0%,使用前不需干燥处理,未对外提供不确定度,因此不考虑对照品纯度引入的不确定度。
(2)对照品称量引入的相对不确定度:①天平的示值误差引入的不确定度:实验所用电子天平的检定证书中,d=0.01 mg时最大允差为±0.1 mg,按矩形分布计算则其标准不确定度:
u(m1)=0.1÷3½=5.77×10-2mg
②天平称量重复性引入的不确定度:电子天平的检定证书中d=0.01 mg时,天平的重复性误差为±0.05 mg,按矩形分布计算其标准不确定度:
u(m2)=0.05÷3½=2.89×10-2mg
③试验称量采用减重法,由相对独立的两次称量组成,因此称量所引入的相对不确定度:
u(m)=2½×﹝u2(m1)+u2(m2)﹞½=9.13×10-2mg
每次称量对照品平行称取两份,盐酸麻黄碱对照品称样量分别为10.26 mg、10.33 mg,其每份对照品称量引入的相对标准不确定度分别为:
urel(m对1)=u(m)÷m对1=9.13×10-2÷10.26=8.90×10-3mg
urel(m对2)=u(m)÷m对2=9.13×10-2÷10.33=8.84×10-3mg
因此,对照品称量引入的相对标准不确定度为:
urel(m对)=2½×﹝urel2(m对1)+urel2(m对2)﹞½=1.77×10-2mg
(3)对照品、供试品稀释引入的相对标准不确定度:①玻璃仪器校准引入的不确定度,实验过程使用了A级100 ml容量瓶、50 ml容量瓶及25 ml移液管。根据国家计量检定规程JJG196-2006规定,其最大允许误差分别为±0.10 ml、±0.05 ml、±0.10 ml。按三角分布计算,玻璃仪器校准引入的标准不确定度分别为:
u(V1)a=0.1÷6½=4.08×10-2ml
u(V2)a=0.05÷6½=2.04×10-2ml
u(V3)a=0.1÷6½=4.08×10-2ml
②温度引入的不确定度,实验时的温度为23℃,校准温度为20℃,水的膨胀系数为2.1×10-4ml/℃,按矩形分布计算,温度引入的标准不确定度分别为:
u(V1)b=(2.1×10-4×100×3)÷3½=3.64×10-2ml
u(V1)b=(2.1×10-4×50×3)÷3½=1.82×10-2ml
u(V1)b=(2.1×10-4×25×3)÷3½=9.1×10-3ml
每次稀释,由玻璃仪器校准和温度引入的相对不确定度分别为:
urel(V1)=﹝u2(V1)a+u2(V1)b﹞½÷100=5.47×10-4
urel(V2)=﹝u2(V2)a+u2(V2)b﹞½÷50=5.47×10-4
urel(V3)=﹝u2(V3)a+u2(V3)b﹞½÷25=1.67×10-3ml
则对照品稀释引入的相对标准不确定度为:
urel(v对)=2½×﹝urel2(v1)+urel2(v2)+urel2(v3)﹞½=2.60×10-3
则供试品稀释引入的相对标准不确定度,即:
urel(v供)=2½×﹝urel2(v3)﹞½=2.36×10-3
(4)对照品色谱峰引入的相对标准不确定度
实验中,盐酸麻黄碱对照品配制两份,每份平行进样两次测定峰面积,对照品溶液1的峰面积分别为760.11,759.74,两者差值R1=0.37,平均峰面积为759.92;对照品溶液2的峰面积分别为759.85,759.64,两者差值R2=0.21,平均峰面积为759.74。采用极差法计算,当n=2时,极差系数C=1.13。则每份对照品溶液色谱峰峰面积引入的相对标准不确定度为:
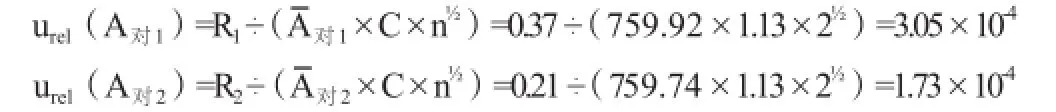
则对照品溶液色谱峰引入的相对标准不确定度为:
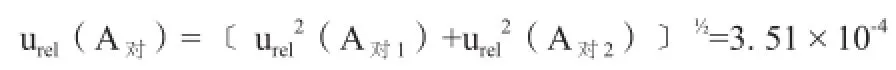
(5)对照品溶液配制的相对标准不确定度:
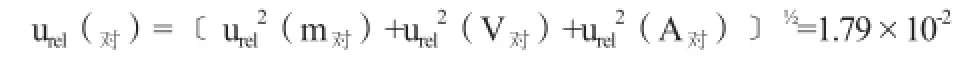
3.2.2 供试品溶液配制和供试品色谱峰引入的不确定度评定:(1) 供试品称量引入的相对不确定度:①天平的示值误差引入的不确定度:所用电子天平的检定证书中,d=0.1 mg时最大允差为±1.0 mg,按矩形分布计算其标准不确定度:
u(m1')=1.0÷3½=5.77×10-1ml
②天平称量重复性引入的不确定度:所用电子天平检定证书中,d=0.1 mg时天平的重复性误差为±0.1 mg,按矩形分布,则其标准不确定度为:
u(m2')=0.1÷3½=5.77×10-2ml
③试验称量采用减重法,由相对独立的两次称量组成,因此称量所引入的相对不确定度:
u(m')=2½×﹝u2(m1')+u2(m2')﹞½=0.82ml
每次称量供试品平行称取两份,称样量分别为0.301 7 g、0.302 4 g。每份供试品称量引入的相对标准不确定度分别为:
urel(m样1)=u(m')÷m样1=0.82÷301.7=2.72×10-3
urel(m样2)=u(m')÷m样2=0.82÷302.4=2.71×10-3
那么供试品称量引入的相对标准不确定度为:
urel(m样)=2½×﹝urel2(m样1)+urel2(m样2)﹞½=5.43×10-3
(2)供试品稀释引入的相对标准不确定度:
urel(V供)=2½×﹝urel2(v3)﹞½=2.36×10-3
(3)供试品色谱峰峰面积引入的相对标准不确定度:实验中,供试品配制两份,每份平行进样两次测定峰面积。供试品溶液1的色谱峰峰面积分别为875.18,877.94,两者差值R3=2.76,平均峰面积为876.56;供试品溶液2的色谱峰峰面积分别为884.16,879.32,两者差值R4=4.84,平均峰面积为 881.74。采用极差法计算,当n=2时,极差系数C=1.13。则每份供试品溶液色谱峰峰面积引入的相对标准不确定度为:
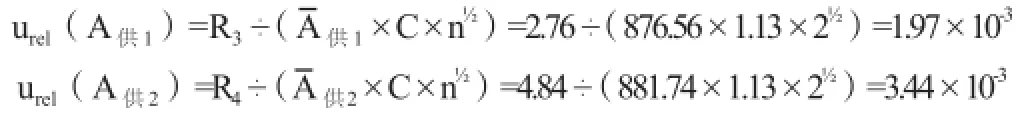
那么,供试品溶液色谱峰峰面积引入的相对标准不确定度为:
urel(A供)=﹝urel2(A供1)+urel2(A供2)﹞½=3.96×10-3
(4)供试品溶液配制的相对标准不确定度
urel(A供)=﹝urel2(m供)+urel
2(V供)+ure
l2(A供)﹞½=7.12×10-3
3.3 合成相对不确定度的评定
苏黄止咳胶囊中盐酸麻黄碱的含量测得结果的合成相对不确定度为:

3.4 扩展不确定度的评定
苏黄止咳胶囊中盐酸麻黄碱的含量测定结果按正态分布,取置信概率P=95%,包含因子k=2,则扩展不确定度为:
U=urel×k=1.93×10-2×2=3.86×10-2
3.5 测量不确定度报告
苏黄止咳胶囊盐酸麻黄碱的含量测定结果,可表示为x=(2.22±0.038 6)mg/粒。苏黄止咳胶囊中盐酸麻黄碱的含量不确定度分量一览表见表1,表中列出了不确定度的因素,及占总不确定度的百分比。
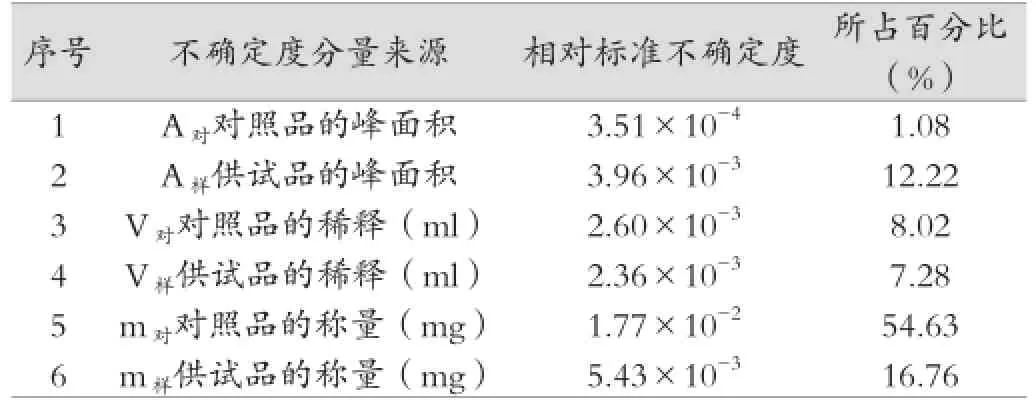
表1 各相对标准不确定度一览表
从表1中可以发现,对照品的称量是苏黄止咳胶囊中盐酸麻黄碱含量测定不确定度的最大影响因素,其次是供试品的称量。
4 讨论
本文评定了苏黄止咳胶囊中盐酸麻黄碱含量测定的扩展不确定度为0.038 6 mg/粒,盐酸麻黄碱含量的测定结果为(2.22± 0.038 6)mg/粒。因此,在药品检验过程中,相关测量设备必须经过计量,且在计量有效期内。检定的量程范围和精度应当涵盖实际称量的范围和测定使用的要求。同时,要建立、健全测量设备的操作、清洁、维修、维护保养工作。定期对测量设备的计量特性是否符合测量过程的计量要求进行确认,尽量减少测量设备本身引入的不确定度分量。
评定不确定度时,合成不确定度的大小取决于各不确定度分量,必须根据实际测量情况进行具体分析,可从测量仪器、测量环境、测量人员、测量方法等方面考虑,尽量做到不遗漏、不重复。苏黄止咳胶囊中盐酸麻黄碱含量测定的评定结果表明,对测量结果的测量不确定度起主要作用的分量来自对照品和供试品的称量过程。测量不确定度的评定,是为了评价测量结果的准确性和可信程度,在药品检验中具有重要意义。
[1]JJF1059. 1-2012测量不确定度评定与表示[S]. 2012.
[2]郑剑红,黄诺嘉. HPLC法测定三黄片中大黄素和大黄酚含量的不确定度评定[J]. 中国医药指南,2010,8(12): 46-48.
[3]赵霞,郭舒岗,谷日旭,等. 高效液相色谱法测定果脯中柠檬黄含量的不确定度分析[J]. 实用医技杂志,2012,19(4): 378-379.
[4]钱江,曹琳,周征. 马来酸氯苯那敏片含量测定的不确定度分析[J]. 西北药学杂志,2015(5): 581-584.
[5]周燕,金佩芬,薛磊冰. HPLC法测定双黄连胶囊中黄答昔黄芩苷含量不确定度评定[J]. 中国药师,2011(2): 19-21.
[6]郑明,金情政,徐辉. HPLC法测定罗红霉素分散片含量的不确定度分析[J]. 海峡药学,2013,25(6): 40-41.
[7]孙莉. HPLC法测定药物含量的不确定度分析[J]. 安徽医药,2010,14(7): 776-777.
[8]王文昭,王路,陈凯. HPLC法测定诺氟沙星胶囊含量的不确定度分析[J]. 中国药事,2010(6): 590-593.
[9]寇光,刘玲,陶松,等. HPLC测定桉叶止咳糖浆中盐酸麻黄碱的不确定度评价[J]. 中国实验方剂学杂志,2013,19(17):41-44.
[10]方海红,陈振华,吴杰连,等. 双波长HPLC测定双黄连口服液中3种成分的不确定度分析[J]. 中国实验方剂学杂志,2012,18(17): 104-108.
[11]陶松,刘旭海,刘玲,等. HPLC测定补肾口服液中淫羊藿苷的不确定度分析[J]. 中国实验方剂学杂志,2012,18(13): 111-114.
[12]吕尚,魏惠珍,饶毅,等. 原子吸收火焰法测定药用辅料山梨醇中的镍元素及其不确定度评价[J]. 中国实验方剂学杂志,2011,17(20): 52-55.
[13]饶毅,夏川川,周海滨,等. 高效液相色谱法测定白芍总苷的不确定度分析[J]. 中国实验方剂学杂志,2011,17(19): 89-93.
[14]王磊,姚令文,程显隆,等. 甘遂中大戟二烯醇HPLC定量方法的测量不确定度评定[J]. 中国药事,2015(2):147-152.
[15]林珍,姚文松,吴娜梅,等. HPLC法测定拉米夫定片剂含量的不确定度评定[J]. 中国药房,2015,26(21): 2968-2970.
[16]杨娟,孙黎,安富荣,等. HPLC测定四草通脉胶囊中槲皮素含量的不确定度评定[J]. 中国药师,2014(12): 2143-2145.
[17]胡丹. HPLC法测定注射用奥沙利铂含量的不确定度分析[J].海峡药学,2014,26(6): 49-51.
[18]成源,李晓雯. HPLC法测定厄贝沙坦片含量的不确定度分析[J].中国药师,2014,(2): 326-328.
Analysis of Uncertainty for Determination of Ephedrine Hydrochloride in Suhuang Zhike Capsule by HPLC
FENG Jie CHENG Qiangbo CHEN Juan CHEN Guohui Beijing Haiyan Pharmaceutical Co.Ltd of Yangtze River Pharmaceutical Group,Beijng 102206,China
ObjectiveTo find out the effect factors of uncertainty by analyzing the measurement uncertainty in the determination of ephedrine hydrochloride in suhuang zhike capsule by HPLC,and provide the evidence for the measurement evaluation.MethordsA mathematic model for calculating uncertainty was established and each component of the uncertainty was assessed.ResultsThe expanded standard measurement uncertainty for the HPLC determination was 0.038 6,and the determined content was(2.22±0.038 6).ConclusionThe established mathematical model of uncertainty measurement is reasonable and reliable. It is applicable to the uncertainty analysis of high performance liquid chromatography method in the determination of drugs.
Suhuang zhike capsule,Ephedrine hydrochloride,HPLC,Uncertainty analysis
R28
B
1674-9316(2015)28-0142-04
10.3969/j.issn.1674-9316.2015.28.108
102206 扬子江药业集团北京海燕药业有限公司
丰杰,E-mail:fengjie@yangzijiang.com
国家“重大新药创制”科技重大专项(2010ZX09401-303-2-6)
猜你喜欢
环球时报(2022-05-18)2022-05-18
中国典型病例大全(2022年9期)2022-04-19
药学实践杂志(2021年6期)2021-12-04
山西中医药大学学报(2020年2期)2020-06-06
中成药(2018年12期)2018-12-29
Asian Journal of Urology(2015年3期)2015-12-16
中国卫生标准管理(2015年14期)2015-01-27
家庭医学(2014年6期)2014-09-11
中国药业(2014年19期)2014-05-17
郑州大学学报(理学版)(2014年2期)2014-03-01
