
金黄色葡萄球菌双组份调节系统SaeRS对重要毒力基因的分子调控机制
2015-12-27 02:28许园园丁宇刘云灵李丹王良兴余方友
温州医科大学学报 2015年9期
许园园,丁宇,刘云灵,李丹,王良兴,余方友
(1.宁波市医疗中心李惠利医院 呼吸内科,浙江 宁波 315041;2.温州医科大学附属第一医院检验科,浙江 温州 325015;3.温州医科大学附属第一医院 呼吸内科,浙江 温州 325015)
·论 著·
金黄色葡萄球菌双组份调节系统SaeRS对重要毒力基因的分子调控机制
许园园1,丁宇2,刘云灵3,李丹2,王良兴3,余方友2
(1.宁波市医疗中心李惠利医院 呼吸内科,浙江 宁波 315041;2.温州医科大学附属第一医院检验科,浙江 温州 325015;3.温州医科大学附属第一医院 呼吸内科,浙江 温州 325015)
目的:初步研究金黄色葡萄球菌双组份调节系统SaeRS对重要毒力基因的分子调控机制。方法:PCR扩增双组份调节系统SaeRS的反应调节蛋白SaeR基因,构建重组表达质粒pET28a-saeR,用限制性酶切、PCR和基因测序方法进行鉴定。用IPTG诱导表达重组蛋白HisSaeR,用镍离子螯合亲和层析法纯化,用Western blot法鉴定。用实时荧光定量RT-PCR方法检测金黄色葡萄球菌SA75及SA75ΔsaeRS突变株luk-PV、hla、coa、fnbB、sak、psmβ基因转录水平。凝胶迁移阻滞实验验证纯化蛋白HisSaeR对这些毒力基因启动区序列的结合能力。结果:重组表达质粒pET28a-saeR构建成功;在0.4 mmol/L IPTG,25 ℃培养12 h的条件下,重组蛋白HisSaeR在大肠杆菌中以可溶形式高效表达;与SA75野生株相比,∆saeRS突变株luk-PV、hla、coa、fnbB基因转录水平均明显下降,分别为野生株的19.7%、0.3%、31.6%、3.5%;psmβ基因和sak基因转录水平无变化。反应调节蛋白SaeR能有效结合luk-PV、hla、coa、fnbB及其自身P1启动区序列。结论:金黄色葡萄球菌双组份调节系统SaeRS对luk-PV、hla、coa、fnbB基因表达具有正调控作用,而这种作用可能是通过反应调节蛋白SaeR与这些基因启动区序列直接结合而实现。
金黄色葡萄球菌;双组份调节系统;saeRS;反应调节蛋白SaeR
金黄色葡萄球菌是引起医院获得性和社区获得性感染的重要病原菌之一,可引起一系列的疾病,包括轻微的皮肤疖痈、创口感染和食物中毒、脓毒性关节炎,以及致命性的、深部的坏死性肺炎、心内膜炎、败血症及多发转移性并发症等[1-3]。金黄色葡萄球菌的高致病性与其产生的多种毒力因子密切相关,包括在生长早期高表达的细胞壁及细胞壁相关蛋白,如纤维蛋白结合蛋白(FnbB)等,和生长晚期高表达的分泌型蛋白,如杀白细胞素(PVL)、α-溶血素(Hla)、血浆凝固酶(Coa)、葡萄球菌激酶(Sak)、β-酚溶调节蛋白(Psmβ)等,这些毒力因子参与细菌的黏附、内化、炎症反应、组织损伤、免疫逃避及细胞毒性等致病过程[1,4]。
金黄色葡萄球菌存在着一个复杂的调节网络,包括sar、sigB、rot等DNA结合蛋白[5]和agr、saeRS、arlRS等双组份调节系统[6-7](two-component signal transduction systems,TCSs),共同调节毒力基因的表达。金黄色葡萄球菌saeRS(staphylococcus aureus exoprotein expression)就是典型的TCSs。saeRS由2个共转录基因saeR和saeS组成,分别编码反应调节蛋白SaeR和组氨酸蛋白激酶SaeS[8]。金黄色葡萄球菌TCRS-SaeRS对多种毒力基因的表达都是必需的,被认为是关键的全局调节子之一。但SaeRS是如何对毒力基因进行调节的分子机制目前还不是很清楚。为此本研究表达纯化双组份中的反应蛋白SaeR,检测saeRS基因敲除后毒力基因的转录水平的变化,同时初步验证SaeR蛋白与疑似靶基因启动子区序列的结合能力。
1 材料和方法
1.1 菌株和质粒 金黄色葡萄球菌临床分离株(SA75)及其saeRS敲除株∆saeRS[9],由本实验室构建并保存(具体的构建方法及敲除株的验证见本课题组先前发表的文章,参考文献[9]。大肠埃希菌E.coli DH5α及BL21(DE3)购于天根生化科技有限公司;表达载体pET28a质粒,购自Novagen公司。
1.2 仪器和试剂 Realplex荧光定量PCR仪(德国Eppendorf公司);超声破碎仪(美国Qsonica公司);电泳仪(美国Bio-Rad公司);限制性内切酶XhoI和BamHI购自Fermentas公司;DNA Marker、琼脂糖凝胶回收试剂盒、基因和质粒组抽提试剂盒、蛋白定量试剂盒、HisTag单抗购自北京天根生化科技有限公司;胰蛋白胨大豆肉汤培养基(TSB)、酵母提取物、胰蛋白胨购自英国Oxiod公司;溶葡萄球菌酶、IPTG购于上海生工有限公司;RNA抽提试剂盒、Ni-NTA agarose购于Qiagen公司;SYBR Premix Ex Taq购于TaKaRa公司;反转录试剂盒购于Bio-Rad公司;DIG Gel Shift Kit购于Roche公司;羊抗鼠IgG-HRP购于Santa cruz公司;引物合成和DNA测序由上海桑尼有限公司完成。
1.3 重组表达质粒pET28a-saeR的构建和鉴定 本实验所有引物均根据NCBI上公布的金黄色葡萄球菌MW2基因组(NC-003923)序列设计,引物序列见表1。以SA75菌株的基因组为模板,saeR-F和saeR-R为引物,扩增saeR基因全长片段;经快速限制性内切酶BamH I和XhoI双酶切后连入pET28a质粒,转入E.coli DH5α,抽提质粒,经酶切、PCR及DNA测序后获得重组表达质粒pET28a-saeR,随后转入表达菌株E.coli BL21(DE3),获得重组表达菌株BL21-pET28a-saeR。抽提重组质粒,进行BamH I、XhoI双酶切鉴定,以saeR-F和saeR-R为引物进行PCR鉴定,最后以pET28质粒自带的T7引物进行重组质粒的基因测序。三项均符合的重组质粒即表明该重组质粒即为成功构建的表达质粒pET28a-saeR。
1.4 重组蛋白HisSaeR的诱导表达和纯化 将重组表达菌株BL21-pET28a-saeR二次活化后,按1∶200比例接种于1 000 mL的Kan+(50 μg/mL)的LB液体培养基中,培养至OD600≈0.6,加入IPTG至终浓度为0.4 mmol/L,25 ℃诱导12 h。离心收集菌体,用PBS洗涤2次。用25 mL非变性结合缓冲液(20 mmol/L咪唑、500 mmol/L NaCl、20 mmol/L Tris、10%甘油,pH 8.0)重悬菌体,冰浴超声破菌(功率200 W,超声5 s,间隔10 s,共2~3 h),4 ℃ 15 000 g离心1 h。收集上清液,加入已平衡好的Ni-NTA agarose亲和层析柱。待样品流近后,加入30 mL的非变性冲洗缓冲液(30 mmol/L咪唑、500 mmol/L NaCl、20 mmol/L Tris、10%甘油,pH 8.0)洗去杂蛋白,最后加入非变性洗脱缓冲液(250 mmol/L咪唑、500 mmol/L NaCl、20 mmol/L Tris、10%甘油,pH 8.0)洗脱目的蛋白。SDS-PAGE分析重组蛋白。
1.5 重组蛋白HisSaeR的Western blot法鉴定 将诱导表达的总蛋白上清,行12% SDS-PAGE电泳后,以恒流转移到PVDF膜上;经5%脱脂奶粉在室温封闭1~2 h,PBST清涤3次;加入鼠源anti-HisTag抗体(1∶1 000)稀释液室温孵育1~2 h,PBST清涤3次;加入羊抗鼠HRP-IgG(1∶2 000)稀释液室温孵育1 h,PBST洗涤3次,最后ECL化学发光显影。
1.6 毒力基因转录水平的Real time PCR技术检测分别收集金黄色葡萄球菌SA75和∆saeRS对数生长中期(3 h)和平台期(9 h)的细菌,用RNA抽提试剂盒提取细菌总RNA,反转录得到cDNA,稀释至浓度为100 ng/μL作为模板,进行Real time PCR,对luk-PV、hla、coa、fnbB、sak、psmβ基因转录水平进行检测,引物序列见表1。其中fnbB基因以生长早期表达量为高,故以3 h的cDNA为模板,而其余基因以平台期表达量最高,故以9 h的cDNA为模板。以管家基因gyrB作为内参,采用相对定量(delta delta Ct,2-∆∆Ct)法计算突变株毒力基因mRNA量相对于野生株的百分率,实验重复3次。

表1 引物序列
1.7 凝胶迁移阻滞实验 以SA75基因组DNA为模板,PCR扩增P1、luk-PV、hla、coa、fnbB启动区序列,引物序列见表1。PCR产物经纯化后用地高辛进行标记。取不同浓度的磷酸化的重组蛋白HisSaeR和0.08 ng标记的启动区序列25 ℃共孵育20 min,同时设立特异性竞争剂(未标记的启动区序列,记为cold)和非特异性竞争剂(未标记的saeR内部序列,记为N)作对照。孵育完后经8%非变性聚丙烯酰胺凝胶电泳,转移至尼龙膜,120 ℃烤膜30 min,马来酸洗液(0.1 mol/L Maleic acid、0.15 mol/L NaCl、0.3% Tween 20,pH 7.5)洗涤3次,用1× Blocking solution室温封闭30 min,马来酸洗液洗涤3次,加入75 mU/mL的Anti-dig-AP抗体稀释液室温孵育30 min,马来酸洗液洗涤3次,随后加入检测缓冲液(0.1 mol/L Tris-Cl、0.1 mol/L NaCl,pH 9.5)平衡5 min,最后用CSPD化学发光显影。
2 结果
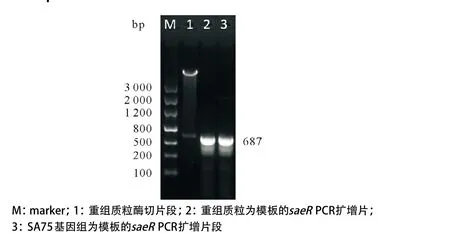
图1 重组表达质粒pET28a-saeR的鉴定
2.1 重组表达质粒pET28a-saeR成功构建 为体外表达金黄色葡萄球菌SA75反应调节蛋白SaeR,以其基因组为模板,PCR扩增出saeR全长(保留终止子,不包括酶切位点,共687 bp);PCR片段经BamH1/ XhoI双酶切后插入表达载体pET28a,得到重组表达质粒pET28a-saeR。重组表达质粒经BamH1/XhoI双酶切及PCR扩增saeR基因可得到理论大小的片段(见图1)。最后DNA测序确认基因序列正确,表明重组质粒pET28a-saeR构建成功。
2.2 重组蛋白HisSaeR的诱导表达 蛋白质若能在上清中表达,可减少实验操作对其自然构型及功能活性的破坏,而温度对表达影响较大,故对诱导温度进行了优化。在细菌生长至OD600为0.6时加入0.4 mmol/L IPTG,之后分别在37 ℃诱导6 h,30 ℃诱导8 h和25 ℃诱导12 h的条件下诱导表达。收集菌体后经超声破碎处理,高速离心后取裂解上清液进行12% SDS-PAGE凝胶电泳。结果显示:诱导后的重组表达菌株BL21-pET28a-saeR的裂解上清液均可见约29 kDa大小的诱导条带,与预测蛋白的分子量一致,且25 ℃诱导 12 h条件下的HisSaeR表达水平较高,而空载菌株BL21-pET28a未见诱导条带(见图2)。
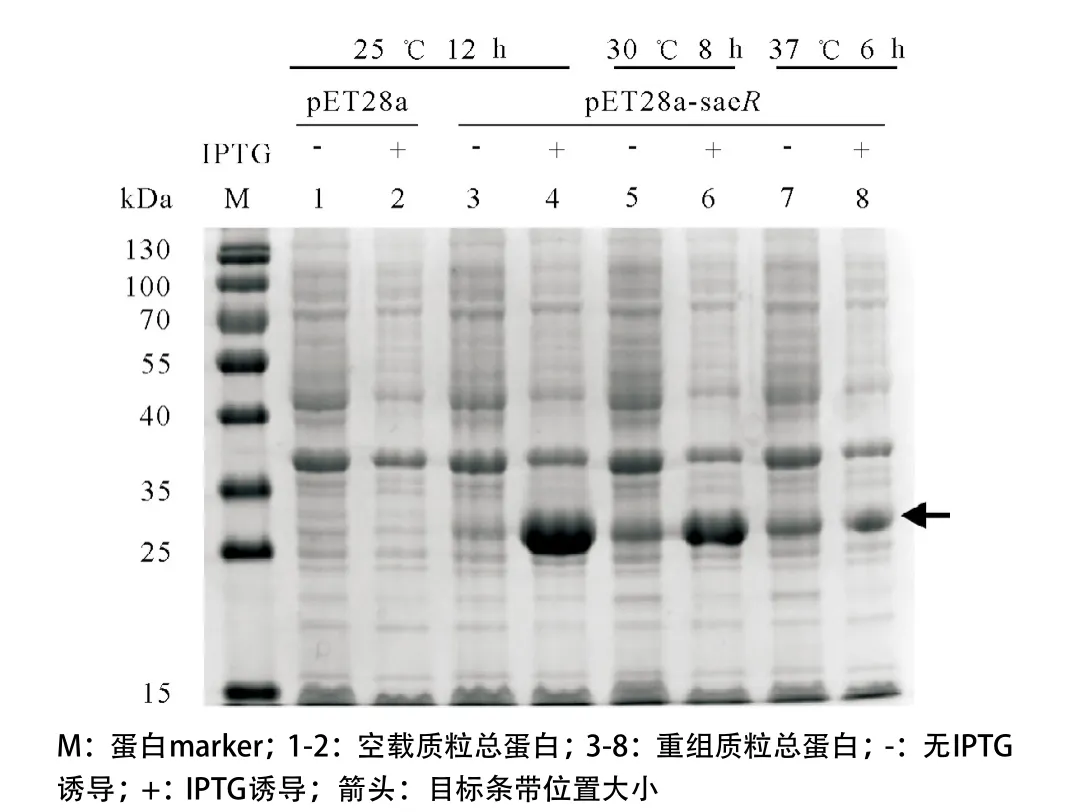
图2 重组蛋白HisSaeR在E.coli BL21中的表达
2.3 重组蛋白HisSaeR的鉴定结果 为鉴定29 kDa大小的蛋白确实为目标蛋白,利用重组蛋白所带的His标签,以anti-HisTag抗体为一抗,采用Western blot法对重组蛋白进行鉴定。结果显示:含重组质粒pET28a-HisSaeR的E.coli BL21的菌体裂解上清中,可检测到信号,而含空载pET28a质粒的E.coli BL21菌体裂解上清中未检测到信号(见图3),从而确认该条带即是目标蛋白。

图3HisSaeR重组蛋白Western blot法鉴定
2.4 重组蛋白HisSaeR的纯化 重组蛋白HisSaeR经Ni-NTA agarose亲和层析柱分离纯化后,SDS-PAGE分析得到相对分子质量约为29 kDa大小的较单一的条带(见图4)。

图4 重组蛋白HisSaeR的纯化
2.5 毒力基因转录水平分析结果 为探索saeRS对毒力基因的分子调控机制,我们选择了两类基因首先进行Real time PCR检测,一类为与黏附、定植及扩散相关的基因,如coa、fnbB、sak;另一类为发挥破坏细胞膜,造成免疫损伤的分泌型的毒力基因,如luk-pv、hla、psmβ。结果显示:与SA75野生株相比,∆saeRS突变株luk-pv、hla、coa、fnbB基因转录水平均明显下降,分别为野生株的19.7%、0.3%、31.6%、3.5%;psmβ基因和sak基因转录水平无变化(见图5)。
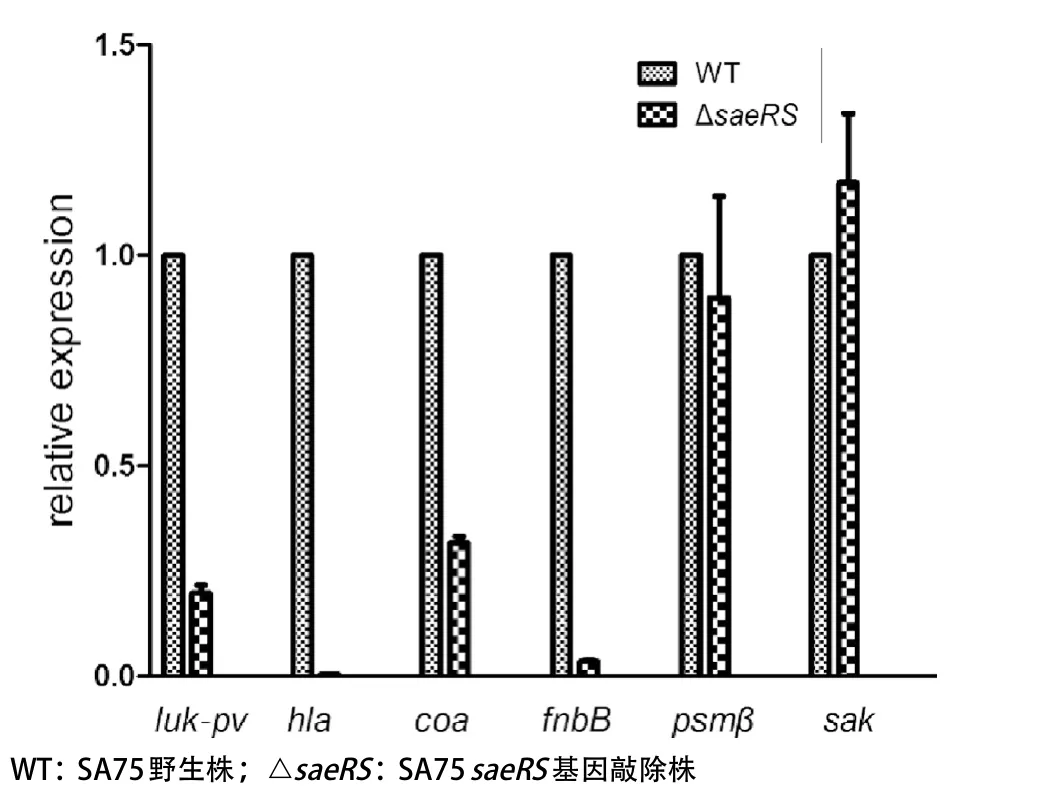
图5 毒力基因转录水平分析
2.6 SaeR蛋白和启动区序列结合能力的凝胶迁移阻滞实验分析结果 根据文献报道,金黄色葡萄球菌磷酸化的SaeR能够与自身P1启动区结合。因此首先进行了HisSaeR和P1启动子的结合实验来验证纯化的蛋白是有活性的。结果显示:与未加蛋白的阴性对照相比,磷酸化的HisSaeR与P1启动区探针孵育后可见迁移阻滞条带,而且随着蛋白浓度的增加,蛋白探针复合物相应增加,阻滞效果越明显,当加入125倍的特异性竞争剂后阻滞效果减弱或消失,而加入125倍非特异性竞争剂后,阻滞条带仍可见(见图6A)。提示磷酸化的重组蛋白HisSaeR能与P1启动区特异结合,并且证明我们的蛋白是有活性的。随后将luk-pv、hla、fnbB、coa这4个基因的可疑启动区序列扩增后dig标记,同样进行EMSA实验。结果显示,磷酸化的SaeR能和这4个基因的启动区序列结合(见图6B-E)。
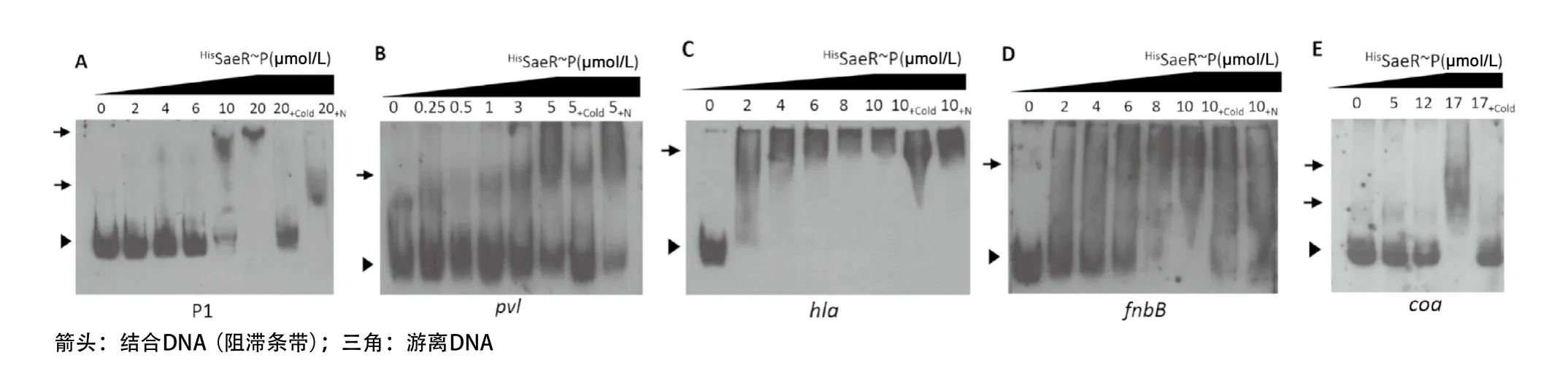
图6 凝胶迁移阻滞实验检测SaeR与毒力基因启动区序列的结合能力
3 讨论
典型的TCSs具有两个组份:一个是位于细胞膜上的组氨酸激酶(histidine kinase,HK)蛋白,能够感受外界信号的变化,使HisKA激酶功能域的组氨酸位点发生自身磷酸化,并将磷酸基团进行传递;另一个是位于细胞质内的反应调节蛋白(response regulator,RR),当接收HK传递的信号后,其N端的天冬氨酸位点发生磷酸化,使其C-末端效应区构象发生改变,暴露不同DNA结合位点,之后与靶基因的启动区结合,发挥转录调节作用[10]。目前研究表明SaeRS双组份就是典型的TCSs,其中SaeR蛋白具有直接和靶基因启动区序列结合的能力。
SaeRS双组份调控系统最早是通过转座子Tn551的插入突变导致表型改变而被发现[11]。sae操纵子由2个启动子(P1和P3)和4个开放阅读框架(saeP、saeQ、saeR和saeS)组成[12],其中saeR和saeS分别编码RR和HK,两者共同组成TCSs。Giraudo等[13]发现将SaeRS突变后凝固酶、α-溶血素、葡萄球菌蛋白A表达明显下降,被认为对毒力的调节具有重要的作用。Rogasch等[14]利用双向电泳技术分析SaeRS突变株的蛋白表达谱,发现其至少可以导致2个细胞表面蛋白和17个胞外分泌蛋白表达下降。Voyich等[15]利用临床菌株MW2(USA400,CA-MRSA)运用基因芯片及动物实验方法对SaeRS的功能进行研究,发现SaeRS影响了212个基因的表达,这些基因不仅涉及细胞毒性,还涉及离子转运、能量代谢及DNA修复等。虽然目前对于金黄色葡萄球菌SaeRS对毒力基因的表达调控作用研究已经较为深入,但SaeRS对毒力基因的分子调节机制目前还不是很明确。较多的研究[16]表明SaeRS双组份调节系统处于调节网络的下游位置,在调节毒力的通路中起到关键的作用,故推测SaeRS对部分毒力基因具有直接调控作用。
因此本实验构建了表达载体pET28a-saeR,纯化了SaeR蛋白;随后用RT-PCR检测了金黄色葡萄球菌SA75和∆saeRS突变株中6个重要毒力基因的表达情况,发现SaeRS对luk-PV、hla、coa、fnbB具有正调控作用,这与上述提到的文献结果相一致;然后用纯化的SaeR蛋白和扩增的毒力基因的启动区序列进行EMSA实验,发现SaeR蛋白能有效结合这些序列。实验结果表明金黄色葡萄球菌双组份调节系统SaeRS对luk-PV、hla、coa、fnbB基因表达的正调控作用是通过SaeR蛋白与这些基因启动区序列直接结合而实现。
总之,本研究构建了重组表达载体pET28a-saeR,成功纯化了具有DNA结合活性的重组蛋白HisSaeR,并初步验证了双组份调节系统SaeRS对部分重要毒力基因的直接调控作用是通过反应调节蛋白SaeR与毒力基因的启动区DNA序列结合而实现,为进一步研究金黄色葡萄球菌TCS-SaeRS调节毒力基因的分子机制打下一定基础。但实验中也存在一些问题,比如SaeR蛋白在金葡菌中的磷酸化是依赖SaeS激活的,而本实验是用化学方法进行的,从而使SaeR蛋白和启动区之间的结合活性不佳,这或许与SaeS在整个结合过程中发挥的功能有关,因此该分子机制的完美诠释还有待进一步研究。
[1] Lowy FD. Staphylococcus aureus infections[J]. N Engl J Med, 1998, 339(8): 520-532.
[2] Archer GL. Staphylococcus aureus: a well-armed pathogen [J]. Clin Infect Dis, 1998, 26(5): 1179-1181.
[3] Foster TJ. The Staphylococcus aureus “superbug”[J]. J ClinInvest, 2004, 114(12): 1693-1696.
[4] Cheung AL, Bayer AS, Zhang G, et al. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus[J]. FEMS Immol/Lunol Med Microbiol, 2004, 40(1): 1-9.
[5] Cheung AL, Zhang G. Global regulation of virulence determinants in Staphylococcus aureus by the SarA protein family[J]. Front Biosci, 2002, 7: d1825-d1842.
[6] Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence[J]. Mol Microbiol, 2003, 48(6): 1429-1449.
[7] Bronner S, Monteil H, Prevost G. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications[J]. FEMS Microbiol Rev, 2004, 28(2): 183-200.
[8] Giraudo AT, Mansilla C, Chan A, et al. Studies on the expression of regulatory locus sae in Staphylococcus aureus[J]. Curr Microbiol, 2003, 46(4): 246-250.
[9] 屠金晶, 张协, 许园园, 等. saeRS对金黄色葡萄球菌临床分离株的hla和lukS-PV表达的影响[J]. 中华微生物学和免疫学杂志, 2013, 33(2): 91-96.
[10] West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems[J]. Trends Biochem Sci, 2001, 26(6): 369-376.
[11] Giraudo AT, Raspanti CG, Calzolari A, et al. Characterization of a Tn551-mutant of Staphylococcus aureus defective in the production of several exoproteins[J]. Can J Microbiol, 1994, 40(8): 677-681.
[12] Steinhuber A, Goerke C, Bayer MG, et al. Molecular architecture of the regulatory Locus sae of Staphylococcus aureus and its impact on expression of virulence factors[J]. J Bacteriol, 2003, 185(21): 6278-6286.
[13] Giraudo AT, Cheung AL, Nagel R. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level[J]. Arch Microbiol, 1997, 168(1): 53-58.
[14] Rogasch K, Ruhmling V, Pane-Farre J, et al. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains[J]. J Bacteriol, 2006, 188(22): 7742-7758.
[15] Voyich JM, Vuong C, Dewald M, et al. The SaeR/S gene regulatory system is essential for innate immol/Lune evasion by Staphylococcus aureus[J]. J Infect Dis, 2009, 199(11): 1698-1706.
[16] Novick RP, Jiang D. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing[J]. Microbiology, 2003, 149(Pt 10): 2709-2717.
(本文编辑:胡苗苗)
Molecular regulatory mechanism of two-component regulatory system SaeRS on important virulencegenes in Staphylococcus aureus
XU Yuanyuan1, DING Yu2, LIU Yunling3, LI Dan2, WANG Liangxing3, YU Fangyou2. 1.Department of Respiratory Medicine, Lihuili Hospital of Ningbo Medical Center, Ningbo, 315041; 2.Department of Clinical Laboratory, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325015; 3.Department of Respiratory Medicine, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325015
Objective: To investigate the molecular regulatory mechanism of two-component regulatory system SaeRS on important virulence genes in Staphylococcus aureus. Methods: The 687-bp saeR gene was PCR amplifed and cloned into pET28a, resulting in the plasmid pET28a-saeR, then tested by restriction enzyme analysis, PCR and gene sequencing. The recombinant proteinHisSaeR was induced by IPTG, purifed with Ni-NTA agarose and verifed by Western blot. The expression levels of luk-PV, hla, coa, fnbB, sak, psmβ genes were detected by real-time PCR. The binding abilities between SaeR and the promoter regions of virulence genes were tested by electrophoretic mobility shift assay. Results: Restriction enzyme digestion, PCR amplifcation and gene sequencing showed that the recombinant plasmid pET28a-saeR was successfully constructed. The recombinant proteinHisSaeR was effciently expressed in soluble in E.coli BL21 (DE3) induced by 0.4 mmol/L IPTG at 25 ℃ for 12 hours. Compared to wild type strain SA75, the 1evels of luk-PV, hla, coa, fnbB mRNA of SA75ΔsaeRS mutant strain decreased to 19.7%, 0.3%, 31.6% and 3.5% respectively whereas the 1evels of psmβ and sak mRNA had no difference between wild type and mutant strain. The electrophoretic mobility shift assay showed that phosphorylated purifedHisSaeR could bind to the promoter regions of luk-PV, hla, coa, fnbB and P1 promoter. Conclusion: Two-component regulatory system SaeRS directly up-regulates the luk-PV, hla, coa, fnbB genes which is realized via response regulator protein SaeR combined with their promoter regions.
Staphylococcus aureus; two-component regulatory system; saeRS; response regulator protein SaeR
R378.1
A
10.3969/j.issn.2095-9400.2015.09.001
2014-12-31
国家自然科学基金资助项目(81271906H2002)。
许园园(1987-),女,浙江宁波人,住院医师,硕士。
余方友,副主任医师,副教授,Email:wzjxyfy@163.com。
猜你喜欢
传染病信息(2022年6期)2023-01-12
现代畜牧科技(2021年9期)2021-10-13
食品安全导刊(2021年20期)2021-08-30
建材发展导向(2021年13期)2021-07-28
农药科学与管理(2019年9期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
创新作文(1-2年级)(2018年2期)2018-09-13
创新作文(小学版)(2018年4期)2018-07-06
学习报·教育研究(2017年33期)2017-08-31
成长·读写月刊(2017年8期)2017-08-12
