
紫胶桐酸合成麝香类大环内酯研究现状
2015-12-24 03:30高山孙彦琳张弘郑华刘兰香
应用化工 2015年1期
高山,孙彦琳,张弘,郑华,刘兰香
(1.昆明理工大学 化学工程学院,云南 昆明 650500;2.中国林业科学研究院 资源昆虫研究所,云南 昆明 650224)
麝香是一种珍贵的香料,是调配高级香精的重要原料之一。从其来源出发可分为天然麝香和合成麝香。由于天然麝香来自鹿科动物成熟雄体香囊的分泌物,资源极其有限,且保护动物的理念愈来愈被人们所接受,所以天然麝香不易获得,因此现在更多地是通过人工合成来弥补其产量的不足[1]。在合成麝香化合物中,大环麝香是三类麝香(硝基麝香、多环麝香及大环麝香)里化学结构最接近天然麝香的一类化合物,因此也最为珍贵。其中,几种常见的大环麝香化合物如图1 所示。图1 中的大环麝香化合物均具有优雅柔和的香型,定香性能良好,接近天然动物类麝香香料的典型气味[2]。

图1 典型大环麝香化合物Fig.1 Typical macrocyclic musk compounds
大环麝香化合物由酮类和内酯类(包括双酯类及醚内酯类)化合物组成,通常大环酮类化合物可采用羟醛缩合法来合成制备,反应条件温和,目前该类化合物的生产已得到较好解决。而大环内酯化合

图2 紫胶桐酸分子结构Fig.2 Molecular structure of aleuritic acid
1 合成方法
合成大环内酯化合物的方法有很多种,根据原料结构的不同,其成环方式也有所区别。从成环的方式而言,主要包括了以酰氧键、碳氧键及碳碳键等成环方式,见表1。

表1 ω-羟基酸合成大环内酯方法Table 1 The synthesis methods of macrocyclic lactone from ω-hydroxy acid
根据表1 所述,由于(5)分子结构中1 位、16 位C 原子所连接的羧基及羟基基团,导致其成环的过程是以形成酰氧键的方式进行的,即活化分子中一端的羧基,使之与另一端的羟基反应生成大环内酯的方法,考虑(5)分子中有三个游离羟基的特殊的分子结构,其合成思路主要有以下两种可供选择。
1.1 邻二醇的保护合成路线
原料(5)分子中的主碳链上接有三个活性较高的羟基基团,都能与分子末端的羧基基团发生反应,同时,在反应过程中分子起始两端一致定位的几率较低,使得合成产物的复杂程度大大增加。因此,一般先将9,10 位邻二醇接上一个环状基团[15]进行保护处理,再进行单分子内酯化作用,最后脱除保护基团得到目标产物。
1976 年,Tseng[16]发明(9E)-异黄葵内酯的合成步骤主要由邻二醇的缩醛保护、脱水成烯以及分子内酯化三步构成(图3)。以Primol 352 作为反应溶剂,对甲苯磺酸催化(5)与原甲酸三甲酯发生缩醛反应得到(6),经乙酸酐脱水消除得(7),再把(7)和KOH 混溶于甘油中通过酯交换生成粗产物,经减压蒸馏[17]获得目标产物(4)。

图3 紫胶桐酸合成(9E)-异黄葵内酯路线1Fig.3 Synthetic route 1 of aleuritic acid to trans-9E-isoambrettolide
在提高反应选择性方面,考虑了(5)分子9,10位的羟基基团也能与羧基基团直接进行酯化反应,导致多种酯类副产物的生成。因此,在大环内酯键连接成键之前,应对该酯化活性位点进行缩醛保护[18],从而降低了副反应的影响,可推知其反应机理见图4。

图4 原甲酸三甲酯与邻二醇缩醛反应机理Fig.4 The alkylation mechanism of trimethyl orthoformate and vicinal diol
该方法优点在于原料(5)与原甲酸三甲酯易得,且反应条件温和,易操作,后续处理只需进行简单的水洗、蒸馏处理即可得到纯度较高、收率高的产物。
吴宪宏等[19]则对上述发明进行了改良,一步合成了目标产物(4),即(5)与原甲酸三甲酯按重量比为1∶(0.5 ~4)投入反应釜后在60 ~180 ℃下完成脱羟基反应;然后加入解聚剂,在真空和180 ~240 ℃下进行解聚环化,得到目标产物(4)。该法不使用质子酸与低碳烷基酸酐催化剂,降低了成本,减少了洗涤污水处理工序,对环境污染的影响大大降低,从而在未来的工业化生产中提供了理论依据。
Venkataraman 等[20-21]发现三聚氯氰(TCT)能够催化长链ω-羟基酸进行内酯化反应(图5)。因此,在溶有(5)的丙酮中先后加入三聚氯氰(TCT)和三乙胺(Et3N),混合于室温条件下反应得到产物(8),即合成(9E)-异黄葵内酯的中间产物。
此反应共分为两个阶段,其一,以丙酮为缩合剂发生缩酮反应,进而避免邻二醇参与后续反应;其二,TCT 催化酯化反应阶段,该反应可能是以三乙胺为诱发剂,(5)末端羧基经TCT 活化生成酰氯,在碱性条件下与羟基发生分子内脱水成酯[22]得到产物(4),而TCT 则转化为二氯羟基-S-三嗪作为不溶物析出。本反应过程通过动力学控制能够使得产物收率高达86%。另外,TCT 其所需反应条件温和,耐水,与大气接触亦可,是一种高效的单分子环化催化剂。

图5 紫胶桐酸合成(9E)-异黄葵内酯路线2Fig.5 Synthetic route 2 of aleuritic acid to trans-9E-isoambrettolide
由于N,N-二甲基甲酰胺二新戊基乙缩醛(DMF-DEA)对高位阻化合物能够显示出独特的优越性,且能与很多含有活泼氢的化合物反应,已经越来越被人们用于醚、硫醚等杂环化合物的合成中。Villemin[23-24]在氮气保护下,将(5)与DMF-DEA 在甲苯混合,发生亲电取代生成(9),然后经醋酸酐脱水、减压蒸馏收集得到油状目标产物(4),其反应过程见图6。

图6 紫胶桐酸合成(9E)-异黄葵内酯路线3Fig.6 Synthetic route 3 of aleuritic acid to trans-9E-isoambrettolide
在(5)邻二醇的保护反应过程中,DMF-DEA 作为反应中间体具有活性高、毒性低、反应条件温和等优点,是合成杂环化合物的理想环化试剂。在DMFDEA 上连有一个电负性较大的二甲氨基基团,使得中心碳原子显正电,羟基进攻碳正离子脱去烷氧基基团,得到较为稳定的环缩醛结构。另外,DMFDEA 不仅具有保护邻二醇的作用,在相同条件下催化羧基酯化得到较高产率的内酯化合物(9),最后醋酸酐催化脱水得到目标产物(4),但是由于(5)分子间的相互偶合使得产物中含有内酯聚合物,最终需要通过碱催化解聚得到内酯单体(4)。
Shiina 等[25-28]在10-樟脑磺酸(CSA)的催化下,(5)与PhCH(OMe)2发生缩醛反应得中间产物(10),经2-甲基-6-硝基苯甲酸酐(MNBA)和4-二甲氨基吡啶氧化物(DMAPO)催化发生内酯化反应获得单体内酯中间产物(11),在醋酸催化下水解得到中间产物(12);最后,被还原的邻二醇与硫代羰基二咪唑反应形成硫代碳酸酯,然后还原脱硫形成卡宾,分子内电子转移脱掉一分子二氧化碳形成双键[29-32],即目标产物(4)。其反应过程见图7。

图7 紫胶桐酸合成(9E)-黄葵内酯路线4Fig.7 Synthetic route 4 of aleuritic acid to trans-9E-isoambrettolide
此方法反应条件温和,在室温条件下即能进行反应。其中,以Et3N 作为缚酸剂,MNBA 为脱水缩合剂,DMAP 作为基质和催化剂,所得产物具有分离纯化操作简便、收率好、纯度高等特点。其原因在于DMAP 吡啶环上的二甲氨基的两个甲基的斥电子效应,一定程度上增加了吡啶环中氮原子的电子云密度,使其偶极矩增大,亲核性明显增强,从而可以进攻羧基碳,达到活化羧基的效果。同时,在MNBA的芳香环上2-位和6-位处引入取代基可以阻止(5)生成其他副产物,这两种效应使得酯化反应活性增强,所合成的中间产物(11)收率好,纯度高。由此推知其反应机理见图8。

图8 DMAP、MNBA 催化羧基酯化反应机理Fig.8 The mechanism of catalyzed esterification reaction by DMAP and MNBA

图9 2-DTC 及DMAP 催化紫胶桐酸衍生物内酯化机理Fig.9 The mechanism of catalyzed lactonization for aleuritic acid derivatives by 2-DTC and DMAP
综合了MNBA 法催化紫胶桐酸环化成酯实验 中存在的缓慢滴加反应物料以及反应时间较长的问题,Oohashi 等[33]利用催化剂碳酸二(2-噻吩)酯(2-DTC)和DMAP 以及I2快速高效地使(10)进行内酯化反应,该法(图9)总共分为两步:首先,原料(10)在2-DTC 和DMAP 的催化下生成中间产物噻吩基酯(13),再在I2的作用下(13)通过电子转移、重排得到中间产物(11)。
1.2 直接合成路线
在目标产物合成过程中,除了上述在原料(5)主碳链上连接环缩醛基团以降低副产物的影响,还可以利用高效催化剂使(5)有选择性地直接进行单分子内酯化反应,从而省去了保护基团进行脱保护处理的中间过程,直接得到目标产物大环内酯。
Venkataraman 等[20-21]为避免丙酮与邻二醇脱水缩合成环,缩短合成过程,则以乙腈为溶剂,三聚氯氰(TCT)催化(5)直接进行酯化环合,经过滤、稀释、碱洗涤,得到产物(12),其反应过程见图10。
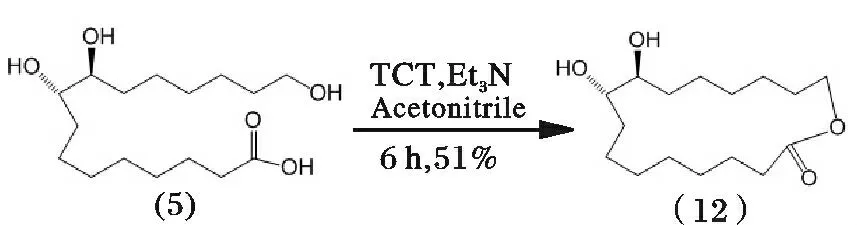
图10 紫胶桐酸合成(9E)-异黄葵内酯路线5Fig.10 Synthetic route 5 of aleuritic acid to trans-9E-isoambrettolide
同时,Shiina 等[25-28]还作了对比试验,直接将(5)一步合成大环内酯粗品,经碱洗涤、干燥、蒸发浓缩及柱层析分离得到单体内酯中间产物(12);然后使用1-硫代羰基二咪唑(TCDI)和三甲氧基膦作用于(12)发生Corey-Winter 反应,并转化为反式烯烃,最终得到目标产物(4),主要反应过程见图11。

图11 紫胶桐酸合成(9E)-黄葵内酯路线6Fig.11 Synthetic route 6 of aleuritic acid to trans-9E-isoambrettolide
2 各合成方法对比
2.1 理论性研究方面
以原甲酸三甲酯和N,N-二甲基甲酰胺二新戊基乙缩醛作为保护基团,经烷酸酸酐催化脱水成烯,经高温解聚而环化的合成方法,虽然合成产物纯度较高,但其合成步骤繁琐,且需在155 ℃的高温条件下进行解聚,能耗消耗较大,反应操作较为繁琐;以TCT 作为催化剂环化合成大环内酯,反应条件温和,室温条件下反应即可,操作简单,但存在目标产物纯度不高且收率低的问题;较2-DTC 催化合成大环内酯的方法而言,虽然以MNBA 催化(5)环化合成大环内酯能够在室温条件下进行反应,且产物收率高达83%等优点,但其反应需要在极稀的物料浓度下进行,反应时间较长,而2-DTC 催化法不仅反应操作简单,而且仅仅在3 h 的反应时间内就能使得目标产物收率达到86%,因此其不失为一种有效的合成方法。
2.2 应用性研究方面
目前以紫胶桐酸为原料合成大环内酯的工业化生产,为数寥寥。然而,在以原甲酸三甲酯和紫胶桐酸为原料一釜合成大环内酯的发明中,其提供的工艺将复杂的反应过程通过简单的工艺步骤在一只反应釜内完成,大大简化了实验操作,从而使得工业化生产得以推广。
3 结束语
由于紫胶桐酸自身结构含有多个反应活性基团,因此在合成大环内酯过程中需要考虑邻二醇的保护基团问题。若使用常规内酯化催化剂时,应在酯化反应前进行预处理,即对9,10 位的羟基进行保护,否则邻二醇的干扰会导致目标产物收率较低;若选择直接催化紫胶桐酸发生内酯化反应时,则需要考虑到紫胶桐酸的自身绕曲闭合是一个熵减过程,从热力学角度是不利的;反应中分子间还存在相互偶合,使反应复杂化。因此,闭环前的核心问题是寻找一种高效催化剂,使紫胶桐酸能够在温和的条件下完成分子单体的闭环过程,且不影响其它已存在的官能团。同时,闭环酯化条件还要满足产率高、纯度好等要求。
在我国,由于我们对合成麝香香料工业的研究起步比较晚,尤其以紫胶桐酸为原料合成麝香类大环内酯化合物的研究还处于起步阶段,大部分相关研究仍集中在反应动力学、热力学及催化机理等基础性研究方面,这与其他一些国家的差距还很大,所以研究大环内酯工业化生产还有很大的研究空间和广阔的市场前景。
[1] 范成有.香料及其应用[M]. 北京:化学工业出版社,1990:259.
[2] Hill J W,Caiothers W H. Studies of polymerization and ring formation. XX. Many-membered cyclic esters[J].Journal of the American Chemical Society,1933,55(12):5031-5039.
[3] 董燕红,王道全,陈馥衡.大环内酯合成方法的新进展[J].合成化学,2000,8(6):493-505.
[4] 陈晓鸣.资源昆虫学研究进展[M].昆明:云南科技出版社,1999:11-13.
[5] 陈晓鸣.紫胶虫培育与紫胶加工[M].北京:中国林业出版社,2008:10-11.
[6] 周铁生,周露,杨祖武. 紫胶桐酸的制备方法:中国,9204516.6[P].1993-04-14.
[7] Nagappayya S K,Galkar V G.Extraction of aleuritic from seedlac and purification by reactive adsorption on functionalized polymers[J].Industrial and Engineering Chemistry Research,2010,49(14):6547-6553.
[8] Keck G E,Sanchez C,Wager C A. Macrolactonization of hydroxyl acids using a polymer bound carbodiimide[J].Tetrahedron Letters,2000,41(45):8673-8676.
[9] Inanaga J,Hirata K,Saeki H,et al.Rapid esterification by means of mixed anhydride and its application to large-ring lactonization[J]. Bulletin of the Chemical Society of Japan,1979,52(7):1989-1993.
[10]White J D,Green N J,Fleming F F. Tin(IV)-catalyzed lactonization of ω-hydroxy trifluoroethyl esters[J]. Tetrahedron Letters,1993,34(22):3515-3518.
[11] Furstner A,Langemann K. Macrocycles by ring-closing metathesis[J].Synthesis,1997,52(7):792-804.
[12]Stille J K,Tanaka M.Intramolecular palladium-catalyzedcyclizetions of esters containing vinyl triflate and vinylstannane groups at the termini.Sythesis of large ring lactones[J]. Journal of the American Chemical Society,1987,109(12):3785-3786.
[13]Nakada M,Kobayashi S,Shibasaki M,et al.The first total synthesis of the antitumor macrolide,rhizoxin[J]. Tetrahedron Letters,1993,34(6):1039-1042.
[14]Mori K,Tomioka H.Pheromone synthesis,CXL.Synthesis of four macrolide phero-mones to define the scope and limitation of enzymatic macrolactonization[J]. European Journal of Organic Chemistry,1992,10:1011-1017.
[15]Kolb H Y,Sharpless K B.A simplified procedure for the stereospecific transformation of 1,2-diols into Epoxides[J].Tetrahedron Letters,1992,48(48):10515-10530.
[16]Tseng C Y.Intermediate in the process for the preparation of trans-Δ9-iso-ambrettolide:US,4014902[P]. 1976-08-09.
[17]Beets M G J. Process of preparing macrocyclic lactones:US,2936310[P].1960-05-10.
[18]Kita Y,Yoshida Y,Mihara S,et al.Efficient pinacol rearrangement mediated by trimethyl orthoformate[J]. Tetrahedron Letters,1997,38(48):8315-8318.
[19]国际香料香精(杭州)有限公司.一种9-环十六烯内酯合成生产工艺:中国,200810162556. 5[P]. 2008-11-25.
[20]Venkataraman K,Wagle D R.Cyanuric chloride,a useful reagent for macrocyclic lactonization[J].Tetrahedron Letters,1980,21(19):1893-1896.
[21]Venkataraman K,Wagle D R.Cyanuric chloride,a useful reagent for converting carboxylic acids into chlorides,esters,amides and peptides[J].Tetrahedron Letters,1979,20(32):3037-3040.
[22]Haval K P,Mhaske S B,Argade N P. Cyanuric chloride:decent dehydrating agent for an exclusive and efficient synthesis of kinetically controlled isomaleimides[J]. Tetrahedron,2006,62(5):937-941.
[23] Villemin D. A simple synthesis of trans-Δ9-isoambrettolide,dihydroambrettolide,and methyl 16-acetoxy-9-hexadecenoate[J].Synthesis,1987(2):154-155.
[24]Uenishi J,Tanaka Y,Kawai N.(Triisopropylsilyl)acetaldehyde acetal as a novel protective group for 1,2-diols[J].Tetrahedron Letters,2006,47(31):5553-5555.
[25] Shiina I,Hashizume M. Synthesis of (9E)-isoambrettolide,amacrocyclic musk compound,using the effective lactonization promoted by symmetric benzoic anhydrides with basic catalysts[J]. Tetrahedron,2006,62(33):7934-7939.
[26]Shiina I,Kubota M,Ibuka R.A novel and efficient macrolactonization of ω-hydroxycarboxylic acids using 2-methyl-6-nitrobenzoic anhydride(MNBA)[J]. Tetrahedron Letters,2002,43(42):7535-7539.
[27] Goncalves S,Nicolas M,Wagner A,et al. Exploring the one-pot acylation of cyclic 1,3-diones with unactivated carboxylic acid[J]. Tetrahedron Letters,2010,51(17):2348-2350.
[28]Lefranc H,Szymoniak J,Delas C,et al.Addition of chiral β-hydroxyl (protected)enol silanes to benzaldehyde dimethyl acetal:access to polypropionate five-carbon stereosequences[J]. Tetrahedron Letters,1999,40(6):1123-1124.
[29]Corey E J,Winter R A E.A new,sterospecific olefin synthesis from 1,2-diols[J].Journal of the American Chemical Society,1963,85(17):2677-2678.
[30]Tichy M,Sicher J.Synthesis and absolute configuration of tricycle(4,4,0,03,8)dec-4-ene(twistene)[J].Tetrahedron Letters,1969,10(53):4609-4613.
[31]Crich D,Paviovic A B,Wink D J. Synthesis of fully-substituted enediynes by the corey-winter reaction[J]. Synthetic Communications,1999,29(3):359-377.
[32]Dudycz L W. Synthesis of 2’,3’-dideoxyuridine bia the corey-winter reaction[J]. Nucleotides and Nucleic Acid,1989,8(1):35-41.
[33]Oohashi Y,Fukumoto K,Mukaiyama T. A new method of the synthesis of carboxylic esters and lactones with di-2-thienyl carbonate(2-DTC)by the promotion of DMAP and Iodine[J]. Bulletin of The Chemical Society of Japan,2005,78:1508-1519.
猜你喜欢
中国药物滥用防治杂志(2022年7期)2022-08-11
天津中医药(2020年10期)2020-12-10
天津中医药(2020年5期)2020-06-01
新课程·下旬(2019年7期)2019-09-17
天然产物研究与开发(2018年8期)2018-09-10
铜仁学院学报(2018年6期)2018-07-05
浙江中西医结合杂志(2016年2期)2016-01-25
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
中国医学科学院学报(2014年6期)2014-03-11
中国药物经济学(2013年4期)2013-06-07
