
临床诊疗限制型心肌病的研究进展
2015-12-16 02:24黄红综述计晓娟审校
中国循环杂志 2015年6期
黄红综述,计晓娟审校
综述
临床诊疗限制型心肌病的研究进展
黄红综述,计晓娟审校
限制型心肌病的临床症状无明显特异性,且易与缩窄性心包炎误诊,其临床诊断比较困难。由于限制型心肌病的预后较差,目前的根治手术为心脏移植,故其早期诊断及早期干预治疗对于防治心力衰竭的发生及进展尤为重要。本文就限制型心肌病的临床诊断、基因诊断及治疗和预后的研究进展进行综述。
限制型心肌病;病因学;临床诊断;治疗
限制型心肌病(restrictive cardiomyopathy,RCM)是心肌间质纤维增生所致心肌僵硬度升高,导致限制性舒张功能障碍,以单侧或双侧心室充盈受限和舒张容量减少,最终导致心力衰竭的心肌病[1,2]。虽然RCM的发病率较低,约占心肌疾病的4.5%,但其预后较差,且常难以与缩窄性心包炎(constrictive pericarditis,CP)鉴别,从而影响治疗方案及对预后的判断。本文就RCM的临床诊疗进展进行简要综述。
1 临床诊断
据文献报道,RCM的发病与地域、种族、性别等因素相关,多发生于热带和温带地区,但未见大规模统计数据[3-5]。我国上海、云南和广西等各地均有散发的病例报道。大多数RCM患者发病年龄较早,男女比例2:1~3:1。目前临床诊断RCM主要根据病史询问、体格检查及辅助检查,包括血液检查、心电图、X线胸片、超声心动图、核磁共振成像、心肌活检、心导管检查等方法。
1.1 临床表现
RCM临床表现差异大,以舒张功能障碍为主,收缩功能正常或接近正常,病程晚期收缩功能可能降低。患者终末期出现右心功能衰竭为主,主要表现为体循环淤血,如颈静脉怒张、肝脏肿大、腹水、下肢水肿、静脉压升高等;部分可出现左心功能衰竭,表现为呼吸困难、咯血及肺底细湿啰音等;也有可能出现低心排出量症状,伴有晕厥,甚至出现血栓栓塞或猝死等。其他的非特异性表现包括乏力、气促、活动耐量减退、体格发育缓慢等[6]。
1.2 诊断及鉴别诊断
RCM临床诊断为排他性诊断[7],需除外肥厚型心肌病[8]、瓣膜性心脏病、心包疾病和先天性心脏病等,主要与CP相鉴别。CP指各种心包炎的最终结局,引起心包粘连增厚,纤维组织增生,致心包纤维化、钙化形成缩窄,继而出现心包增厚和僵硬,心脏活动受限,影响心脏舒张期充盈和容量,从而引起相应的循环系统症状。虽然RCM和CP均表现为舒张功能障碍性心脏疾病,临床症状极其相似,但是两种疾病的治疗方案和预后却有很大差异[9],因此准确诊断对于指导临床治疗有重要的意义。表1为复习文献后总结出的RCM和CP的临床鉴别诊断要点。
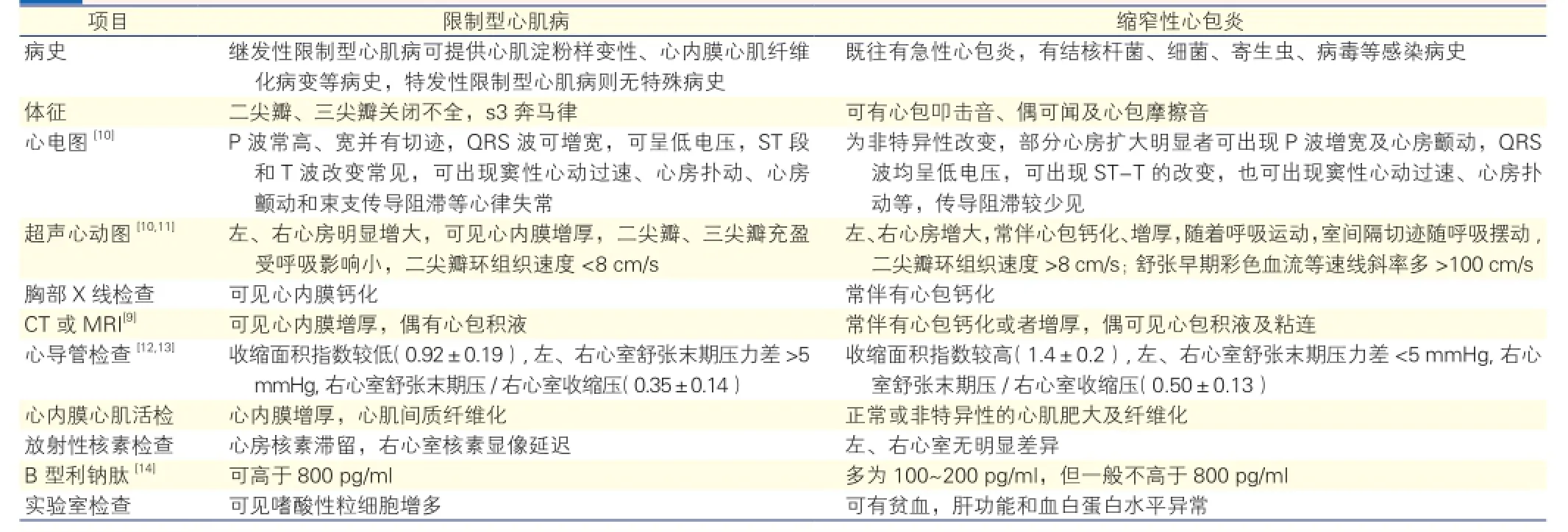
表1 限制型心肌病和缩窄性心包炎的临床鉴别诊断要点
虽然RCM与CP可从不同的方面进行鉴别,影像学检查方法也在不断更新,但临床上因两种疾病无特异性的鉴别点,且临床表现和血液动力学改变极为相似,因此无创性检查常不能完全鉴别这两种疾病[15],这对RCM的临床诊疗提出了巨大的挑战。
据国内外文献报道[15,16],RCM可被误诊为左心室心肌致密化不全、扩张型心肌病、病毒性心肌炎、CP等,其中CP与RCM在临床上最难以鉴别。误诊病例多因劳力性心悸、气促、肝脾肿大等心功能衰竭症状入院。CP患儿误诊为RCM者,多表现为心功能衰竭症状,予以利尿、强心等对症支持治疗效果不佳,最终需通过心脏计算机断层摄影术(CT)、超声心动图等一系列影像学检查鉴别有无心包病变。CP患儿经过心包剥脱术治疗后,多数在症状改善后出院。有文献报道,有病例因影像学鉴别困难未能有效鉴别RCM与CP,但临床诊断为CP而行手术,术中却明确诊断为RCM,最后死于术后并发症[16,17]。由此可见,RCM与CP的临床鉴别比较困难,严重时可因错过治疗时机而影响预后,此时可考虑基因诊断或心肌细胞活检。
2 病因诊断
2008年,欧洲心脏病学会提出,当临床难以明确诊断心肌病时,应注重心肌病的病因诊断,针对病因进行探索性治疗[2]。RCM按照病因分类,分为特发性RCM和继发性RCM。
2.1 特发性限制型心肌病病因
特发性RCM多见于儿童,且目前大量文献提示基因突变为其主要病因[18],表2为目前已知的RCM相关突变基因。

表2 限制型心肌病心肌蛋白基因突变报告位点
目前国内、外的研究热点为心肌蛋白相关基因突变,研究大致有三类。
肌钙蛋白I(TNNI)的基因错义突变:Yumoto 等[25]研究TNNI3基因突变可致心肌细胞Ca2+的敏感度改变,心肌收缩力增强和舒张功能受损。国内外研究者发现,RCM患者TNNI3突变位点所致结果包括TNNI3-8 Asp190His、Arg192 His、TNNI3-7 Lys178Glu、Arg145Trp、Ala171Thr、Leu144Gln,均为单基因突变位点。2013年发表的3例限制型心肌病患儿遗传分析结果显示,其中2个病例分别发现了TNNI3基因突变位点,分别为Arg204 His和Arg192 His,研究表明这样的基因突变可引起心肌纤维对Ca2+的敏感度增加,造成心肌收缩力增强和纤维舒张功能受损[23]。
肌钙蛋白T(TNNT)的基因缺失突变:国内学者杨世伟等[23]对1例限制型心肌病患儿排除继发性心肌病后,行遗传基因分析发现,TNNT2基因第9外显子新发双缺失突变(c.297-302 AATGAG deletion),造成第100.101号相邻编码氨基酸的缺失(p.Asn100 Glu101 del)。该研究显示,Asn100单个缺失可造成心肌细胞Ca2+的敏感度增加,心肌收缩力增强和纤维舒张功能受损,Glu101单个缺失可造成心肌细胞Ca2+的敏感度降低,而p.Asnl00-Glu10l del双缺失的整体效应表现为:心肌细胞Ca2+的敏感度增加,心肌收缩力增强和舒张功能受损。Pinto[24,26]发现,TNNT2基因突变可影响心肌Ca2+的敏感度。
结蛋白基因杂合突变:结蛋白为存在于心肌、骨骼肌、平滑肌中的中间丝蛋白。结蛋白相关研究提示,结蛋白基因突变可致各种心肌病。有文献报道,临床上确诊RCM者检测出结蛋白外显子基因突变,而该突变可导致结蛋白Glu413Lys[20]。
基因突变的临床意义:由于表观遗传学作用,编码心肌相关蛋白的基因突变可致不同种类的原发性心肌病,包括MYH7、MYBPC3、TNNT2突变;MYH7、MYBPC3突变可见于RCM,也可见于肥厚型心肌病。但上述基因突变位点均未在其他疾病中发现。
由于心肌病发病率较低,尚缺乏大规模、多中心临床数据证实基因突变位点与临床疾病的直接关系。对于RCM的诊断,依旧需要结合临床诊断,基因诊断可以提供病因学资料。
2.2 继发性限制型心肌病病因
继发性RCM多继发于全身系统疾病,如浸润性或贮积性疾病(淀粉样变[8]、结节病、血色病) 累及心肌;心内膜纤维化、嗜酸细胞性心内膜炎、心内膜纤维弹力增生症累及心内膜;放射线损害,蒽环类抗肿瘤药物累及心肌和心内膜同时受损[2]等。
3 预后
在所有类型心肌病中,RCM预后最差,50%的患者在确诊后2年左右死亡。有研究显示,乌干达死于心力衰竭的患者中,14%左右被诊断为心内膜心肌纤维化[28]。在热带及温热带,RCM的死亡率可达15%~20%。澳大利亚有报告称,所有心肌病中RCM占2.5%[3];另外有资料显示,中国上海RCM发病率约4.7%,随访6个月时死亡率约31.3%[29];郑州相关调查显示,当地RCM发病率为10.8%,死亡率达37.5%[30];Matsumori等[31]报道,RCM发病率为0.2/100 000,年死亡率约为10%,其中1/3死因为猝死或心律失常。
4 治疗
目前为止,RCM的治疗方法主要为对症治疗,包括药物治疗、手术治疗,目的是改善心功能。目前RCM病因研究也是研究重点之一,考虑到基因突变为主要病因,基因治疗或将成为根治RCM的新方向[32]。
药物治疗:RCM药物治疗的原则是改善心室的顺应性、增加心室的充盈和改善舒张功能,但疗效均不满意。药物治疗包括利尿剂、血管扩张剂、钙拮抗剂、心肌营养药物等综合治疗[6]。利尿剂和血管扩张剂可缓解症状,但应注意小剂量使用,避免降低心室充盈而影响心排出量;钙拮抗剂对改善心室顺应性可能有效;舒张功能损害明显者,在发生快速心房颤动时可应用洋地黄制剂改善心室充盈;有附壁血栓和(或)已发生血栓栓塞者应加用抗凝及抗血小板药物。
起搏器治疗:因RCM可致循环衰竭及心律失常等表现,
对于有明显心肌缺血及晕厥的患者,可考虑置入埋藏式心脏复律除颤器,对猝死高危患者有利。
外科手术:(1)心内膜剥脱术:当内科治疗疗效不佳时,提示心内膜纤维化严重病变时,则可选择心内膜剥离术,若累及心脏瓣膜病变,则手术内容包含切除附壁血栓和纤维化的心内膜,置换二尖瓣与三尖瓣。(2)心脏移植:在克服伦理学及心脏排异等难关后,对于RCM患者,心脏移植被视为最为有效的根治术,且已有手术成功的病例[33]。
基因治疗:虽然RCM与多种基因突变有关,但目前尚少见基因治疗的相关报道。不过,针对心肌病的基因治疗已经开始进行有益的探索[34],这将是未来RCM治疗的研究热点。
[1] Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation, 2006, 113: 1807-1816.
[2] Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J, 2008, 29: 270-276.
[3] Nugent AW, Daubeney PE, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med, 2003, 348: 1639-1646.
[4] Sliwa K, Mayosi BM. Recent advances in the epidemiology, pathogenesis and prognosis of acute heart failure and cardiomyopathy in Africa. Heart, 2013, 99: 1317-1322.
[5] Russo LM, Webber SA. Idiopathic restrictive cardiomyopathy in children. Heart, 2005, 91: 1199-1202.
[6] Rivenes SM, Kearney DL, Smith EO, et al. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation, 2000, 102: 876-882.
[7] Merlo M, Abate E, Pinamonti B, et al. Restrictive cardiomyopathy: clinical assessment and imaging in diagnosis and patient management. Clinical echocardiography and other imaging techniques in cardiomyopathies. Germany: Springer International Publishing, 2014. 185-206.
[8] 郭然, 王珂, 郑晓群, 等. 以体循环淤血为主要表现的淀粉样变性心肌病临床特点分析. 中国循环杂志, 2005, 20: 283-284.
[9] Mookadam F, Jiamsripong P, Raslan SF, et al. Constrictive pericarditis and restrictive cardiomyopathy in the modern era. Future cardiology, 2011, 7: 471-483.
[10] 吴炜, 张抒扬, 严晓伟, 等. 家族性限制型心肌病临床特点分析.中国循环杂志, 2013, 3: 203-206.
[11] Welch TD, Ling LH, Espinosa RE, et al. Echocardiographic diagnosis of constrictive pericarditis Mayo Clinic criteria. Circulation: Cardiovascular Imaging, 2014, 7: 526-534.
[12] Denfield SW, Rosenthal G, Gajarski RJ, et al. Restrictive cardiomyopathies in childhood. Etiologies and natural history. Tex Heart I J, 1997, 24: 38.
[13] Talreja DR, Nishimura RA, Oh JK, et al. Constrictive pericarditis in the modern era: novel criteria for diagnosis in the cardiac catheterization laboratory. J Am Coll Cardiol, 2008, 51: 315-319.
[14] Leya FS, Arab D, Joyal D, et al. The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. J Am Coll Cardiol, 2005, 45: 1900-1902.
[15] Chinnaiyan KM, Leff CB, Marsalese DL. Constrictive pericarditis versus restrictive cardiomyopathy: challenges in diagnosis and management. Cardiol Rev, 2004, 12: 314-320.
[16] 刘瑛, 刘东海, 王秀英, 等. 限制型心肌病 7 例临床误诊分析. 临床和实验医学杂志, 2009, 8: 35-36.
[17] 孙贵斌, 金京浩. 缩窄性心包炎与限制型心肌病的临床鉴别: 附2例误诊报告. 工企医刊, 1990, 1: 19-21.
[18] Towbin JA. Inherited cardiomyopathies. Jpn Circ J, 2014, 78: 2347-2356.
[19] Kaski JP, Syrris P, Burch M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart, 2008, 94: 1478-1484.
[20] Pruszczyk P, Kostera-Pruszczyk A, Shatunov A, et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int J Cardiol, 2007, 117: 244-253.
[21] Ware SM, Quinn ME, Ballard ET, et al. Pediatric restrictive cardiomyopathy associated with a mutation in β-myosin heavy chain. Clin Genet, 2008, 73: 165-170.
[22] Purevjav E, Arimura T, Augustin S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet, 2012, 21: 2039-2053.
[23] 杨世伟, 陈彦, 李军, 等. 儿童原发性限制型心肌病三例的临床特征及遗传分析. 中华心血管病杂志, 2013, 41: 304-309.
[24] Pinto JR, Yang SW, Hitz MP, et al. Fetal Cardiac troponin isoforms rescue the increased Ca2+sensitivity produced by a novel double deletion in cardiac troponin T linked to restrictive cardiomyopathy : a clinical, genetic, and functional approach. J Biol Chem, 2011, 286: 20901-20912.
[25] Yumoto F, Lu QW, Morimoto S, et al. Drastic Ca2+sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem Biophys Res Commun, 2005, 338: 1519-1526.
[26] Pinto JR, Parvatiyar MS, Jones MA, et al. A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+sensitivity of force development and impairs the inhibitory properties of troponin. J Biol Chem, 2008, 283: 2156-2166.
[27] Peled Y, Gramlich M, Yoskovitz G, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol, 2014, 171: 24-30.
[28] 葛丽华, 刘梅林, 胡大一. 发展中国家心衰人群的流行病学特点.中华实用医药杂志, 2003, 3: 1576-1578.
[29] 章旭, 黄美蓉, 陈树宝, 等. 儿童心肌病诊断治疗 10 年回顾. 临床儿科杂志, 2011, 29: 345-348.
[30] 董燕, 李艳. 儿童心肌病诊治回顾分析. 医药与保健, 2014, 22: 87.
[31] Matsumori A, Furukawa Y, Hasegawa K, et al. Epidemiologic and clinical characteristics of cardiomyopathies in Japan. Results from nationwide surveys. Circ J, 2002, 66: 323-336.
[32] Biswas A, Rao VR, Seth S, et al. Next generation sequencing in cardiomyopathy: towards personalized genomics and medicine. Mol Biol Rep, 2014:41:4881-4888.
[33] Fenton MJ, Chubb H, McMahon AM, et al. Heart and heart-lung transplantation for idiopathic restrictive cardiomyopathy in children. Heart, 2006, 92: 85-89.
[34] Mearini G, Stimpel D, Geertz B, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun, 2014, 5: 5515.
2015-02-09)
(编辑:朱柳媛)
国家自然科学基金项目(81301300);重庆医科大学附属儿童医院疑难专项研究(hjyn2013-7)
400014 重庆市,重庆医科大学附属儿童医院 心脏中心 儿童发育疾病研究教育部重点实验室 儿科学重庆市重点实验室重庆市儿童发育重大疾病治疗与预防国际科技总基地
黄红 硕士研究生 主要研究方向为儿童心血管疾病诊疗 Email:hongh1029@163.com 通讯作者:计晓娟 Email:jixiaojuan2003@163.com
R54
A
1000-3614(2015)06-0594-03
10.3969/j.issn.1000-3614.2015.06.021
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
协和医学杂志(2021年3期)2021-06-02
中国生殖健康(2020年2期)2021-01-18
中国临床医学影像杂志(2019年1期)2019-04-25
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国医疗设备(2017年12期)2017-12-23
中西医结合心血管病杂志(电子版)(2015年35期)2015-10-25
中国当代医药(2015年31期)2015-03-01
中国实验诊断学(2015年7期)2015-02-24
