
不同形貌稀土掺杂Gd2O3粉体的制备及光磁性能研究
2015-12-15 07:18徐德康刘楚枫阎佳薇欧阳红群张曰理
无机化学学报 2015年4期
徐德康 刘楚枫 阎佳薇 欧阳红群 张曰理
(中山大学物理科学与工程技术学院,广州510275)
不同形貌稀土掺杂Gd2O3粉体的制备及光磁性能研究
徐德康 刘楚枫 阎佳薇 欧阳红群 张曰理*
(中山大学物理科学与工程技术学院,广州510275)
采用水热法,利用不同添加剂:柠檬酸(CA)、油酸(OA)、乙二胺四乙酸(EDTA)制备出了不同形貌前驱物Gd(OH)3,并经退火得到不同形貌的Gd2O3样品(S-CA、S-OA、S-EDTA)。XRD图表明所制得Gd2O3粉末均为立方晶系(空间群为Ia3),并且不同添加剂所得粉晶的晶格常数略有不同:1.082 25 nm(S-CA),1.081 14 nm(S-OA),1.083 20 nm(S-EDTA);SEM图可看出其颗粒大小分别约为63 nm(S-CA),300 nm(S-OA),2 μm(S-EDTA);红外光谱则进一步证明3种产物均为Gd2O3,并且不同添加剂下样品的基团振动吸收强度不一样;利用荧光光谱仪和综合物性测量系统测量研究了经不同添加剂所制备出的稀土(Yb,Er/Ho)掺杂Gd2O3的上转换发光特性及磁学性能,结果表明:样品形貌对稀土掺杂Gd2O3上转换发光强度和顺磁磁化率影响较大,其中由EDTA添加剂所制备出的稀土掺杂Gd2O3粉末的上转换发光强度和顺磁磁化率最佳。
稀土氧化物;添加剂;形貌;发光;磁性
Gd2O3材料由于具有卓越的物理化学稳定性,无毒性以及较高的剩余磁化率,在核磁共振成像[1-2],半导体制造[3-4]等领域具有重要应用。此外,Gd2O3材料的声子频率较低(截止频率低于600 cm-1),允许杂质掺杂浓度较高,在上/下转换荧光基质材料上具有广阔的应用前景[5-8]。制备Gd2O3的方法主要有热解法[9],激光刻蚀法[10],煅烧法[11]等。热解法由于操作简单,成本低廉,并且可以在过程中加入添加剂控制颗粒形貌,因此已成为制备稀土氧化物常用方法。在稀土发光材料中,样品形貌对于稀土发光特性具有重要影响[12-13];然而由于Gd2O3制备方法的限制,难以在制备过程中通过条件控制直接得到均匀的不同形貌的样品。因此,如何有效控制合成不同形貌Gd2O3样品并研究形貌对光磁特性是一个亟需解决的问题。
近年来,通过改变前驱物的形貌,进而退火后得到不同形貌的氧化物得到广泛的关注。例如,中科院长春应化所的林君小组[14]用氨水作OH-源通过水热法合成纳米棒状Gd(OH)3和Gd2O3;时隔两年,应化所的尤洪鹏小组[5]亦用氨水在常温下制备出管状Gd(OH)3和Gd2O3;高能物理所的赵宇亮小组[15]则通过加入不同剂量的丙三醇,得到一系列尺寸的Gd2O3颗粒,并研究其在核磁共振照影剂方面的应用;最近,北京石油化工学院的邢光建小组[16]用葡萄糖添加剂制备出具有高度均匀的Gd(OH)3和Gd2O3六角形形貌的微米颗粒,并讨论其生长机制和荧光性质。另外,Yb3+-Er3+/Ho3+共掺上转换体系一直以来受到广泛地研究,其以Er3+/Ho3+作为激活剂,Yb3+作为敏化剂的能量传递过程无论是在生物探针[17-18],温度感应[19],光学通讯[20],以及光伏技术[21-22]等均有广阔的应用前景。
然而迄今为止,系统地采用不同的添加剂来制备前驱物从而制造出具备不同形貌的Gd2O3及其相关光磁特性的研究尚未见报道,因此本文通过水热以及热处理方法,研究不同添加剂(柠檬酸、油酸、EDTA)对Gd2O3颗粒形貌的影响,进而测量研究不同添加剂对稀土掺杂(Yb3+,Er3+/Ho3+)Gd2O3发光性能和磁学性能的影响,探讨其在光/磁双模探针方面的潜在应用。
1 实验方法
1.1 实验试剂
稀土氧化物(99.9%,Aladdin Chemistry公司)、KOH(85%,广州化学试剂厂)、一水柠檬酸(99.5%,广州润捷化学试剂有限公司)、油酸(分析纯,天津市富宇精细化工有限公司)、乙二胺四乙酸(分析纯,天津市福晨化学试剂仓)、浓硝酸(65%,广州化学试剂厂)。
1.2 实验步骤
分别称量1 mmol Gd2O3,溶于过量浓硝酸后水浴蒸发至结晶,再加入5 mL去离子水形成Gd(NO3)3溶液,并加入18 mmol的KOH搅拌;将上述溶液制成3份样品,分别加入不同的添加剂(0.211 2 g柠檬酸,5 mL油酸或者0.439 7 g EDTA),持续搅拌30 min,得到白色絮状物。控制最终溶液体积为30 mL并转移入聚四氟乙烯衬底,放入高压反应釜在200℃保温12 h,然后降至室温,取出混合液,经高速离心,干燥后得到前驱物Gd(OH)3,分别标记为P-CA、P-OA、P-EDTA,最后经马弗炉在700℃退火后得到Gd2O3粉末,分别标记样品为S-CA,S-OA,S-EDTA。稀土掺杂(Yb3+,Er3+/Ho3+)Gd2O3颗粒粉末的制备与以上无掺杂制备过程完全相同。
1.3 表征方法
前驱物和最终产物的结构由JEOL公司DMax2200 VPC的X射线衍射仪(Cu Kα,λ=0.154 06 nm)进行分析。样品的形貌则由型号为JEOL公司JSM6060的扫描电镜进行观察。红外光谱则由Bruker公司的EQUINOX 55傅里叶变换红外光谱-红外显微镜联用仪进行表征。上转换荧光谱则由搭配了2 W的980 nm激光器的Edinburgh FLS920组合式荧光寿命与稳态荧光光谱仪所得。磁学性能则由Quantum Design综合物性测量系统进行表征。
2 结果与讨论
2.1 结构表征
由材料的X射线衍射图可以看到所制备的Gd(OH)3前驱物为六方晶系(空间群是P6)结构(见图1)。对比六方相Gd(OH)3标准卡片No.38-1042,P-CA和P-OA样品均为纯相,而S-EDTA的则含有些许杂相。另外,由图1可以看出:3种不同添加剂所得Gd(OH)3材料的衍射峰强度略有不同,说明不同添加剂对Gd(OH)3粉末的生长取向有不同的影响,从而导致不同形貌。
图2为经过退火处理后的Gd2O3的XRD图,所制备的Gd2O3为立方晶系结构。同样地,不同添加剂对Gd2O3晶格结构没有产生影响。对比立方晶系Gd2O3衍射标准卡片No.12-0797,在XRD谱线中未发现杂峰,说明在不同添加剂作用下退火所得Gd2O3均为纯相,并且S-EDTA前驱物的杂质对最终产物没有影响。利用Jade软件对样品的XRD衍射数据进行分析可得到3种不同形貌Gd2O3的晶格常数分别为:1.082 25 nm(S-CA),1.081 14 nm(SOA),1.083 20 nm(S-EDTA)。

图1 不同添加剂下Gd(OH)3的XRD图及Gd(OH)3标准卡片No.38-1042Fig.1 XRD patterns for Gd(OH)3samples with different additives and the standard data(No.38-1042)for Gd(OH)3as reference data

图2 不同添加剂下Gd2O3的XRD图及Gd2O3标准卡片No.12-0797Fig.2 XRD patterns for Gd2O3samp les derived from different additives and the standard data (No.12-0797)for Gd2O3as reference data
另外,我们也表征了Yb3+-Er3+/Ho3+共掺的Gd2O3粉末的XRD图,发现其结构亦与未掺杂的Gd2O3相同,均为立方晶系,只是衍射角均发生右移,这可能是由于更小离子半径(rYb=86.8 pm,rEr=89 pm,rHo= 90.1 pm)的镧系元素取代更大离子半径Gd3+(rGd= 93.5 pm)的缘故。
2.2 形貌表征
图3给出了由3种不同添加剂所制备的Gd (OH)3样品的扫描电镜照片。图3(a)为经柠檬酸(CA)添加剂所制备的Gd(OH)3样品的SEM图,可以看出:颗粒基本为球形,颗粒大小分布在70 nm左右。图3(b)为经油酸(OA)添加剂所制备的Gd(OH)3样品的SEM图,由图可以看出:样品成块材级别的颗粒,这是因为在水热反应中Gd(OH)3颗粒间由油酸分子联接起来形成严重团聚,样品分散性很差。图3 (c)则为添加乙二胺四乙酸(EDTA)时样品的SEM图,从图可以看出:样品为规则的六棱柱(见插图),这是因为Gd(OH)3六方相结构在晶体各向异性生长过程中容易形成六棱柱结构,棱柱大小基本一致,棱柱宽度约为1.5 μm,长度约为2 μm。

图3不同添加剂下Gd(OH)3的SEM照片Fig.3 SEM images of the Gd(OH)3samples with different additives
图4 为经700℃退火处理后所得Gd2O3样品的SEM图。由图4(a)可以看出:经CA添加剂作用和退火处理后的Gd2O3颗粒仍为球状结构,其颗粒的大小分布同样在70 nm左右。图4(b)为OA作用下的Gd2O3形貌图,从形貌上看出:退火处理后的Gd2O3颗粒不再团聚起来,但仍为不规则颗粒嵌套而成,整体大小约为300 nm。图4(c)所展示的为经EDTA添加剂作用和退火处理后的Gd2O3颗粒的SEM形貌图,由图可以看出:此种情况所得Gd2O3颗粒形状与其前驱物母体类似,但表面则较为粗糙,可能是在退火过程中结晶度增加以及脱水等原因造成的,颗粒长度约为2 μm。

图4 不同添加剂下Gd2O3的SEM照片Fig.4 SEM images of the synthesized Gd2O3samplesderived from different additives
对比图3和图4可以发现,最终产物的形貌与前驱物的形貌基本相同(油酸所在体系的团聚现象严重,无法获取油酸为添加剂时前驱物的形貌),这说明可以通过调控前驱物的形貌进一步控制产物形貌。而在直接制出Gd2O3的方法中如高温固相等是无法加入添加剂控制形貌的,也就较难控制Gd2O3的形貌。本实验通过对于前驱物形貌的控制实现了对最终产物形貌的调控。另外,Yb3+,Er3+/Ho3+共掺Gd2O3粉末的形貌也与未掺杂的类似,在不同的螯合剂下(CA,OA以及EDTA)的形貌均表现为纳米颗粒,纳米颗粒团聚物,以及六棱柱状颗粒的形貌,尺寸也与未掺杂的相同。
我们也表征了退火后的产物的红外光谱,如图5所示。不同添加剂作用后并经过退火,得到红外光谱的各种基团吸收峰基本一致。从图上可看出,代表Gd2O3的Gd=O振动峰(543.8 cm-1)[23]在3个样品的吸收强度均相同,表明3个样品均为Gd2O3;而最强的双峰(1 382.7和1 500.4 cm-1)是由C=O不对称伸缩振动造成[24],这个可能来自于微/纳米颗粒对空气中碳化物气体的化学吸附导致[25];在3374.9 cm-1的宽峰则是由于颗粒与空气中的水分接触产生的-OH伸缩振动[24]。最后,我们可以看出,不同基团的吸收强度也有规律,强度随着不同添加剂有所变化,其大小表现为:S-CA最大,S-OA次之,S-EDTA最小。

图5 不同添加剂下Gd2O3的IR光谱Fig.5 IR spectrum of Gd2O3samples derived from different additives
2.3 上转换光谱测量
在980 nm的近红外激光激发下,3种Gd2O3∶Yb3+,Er3+呈现肉眼可见的红色荧光。由图6(a)可知,其发光峰位主要为665 nm,表现为红光波段,对应于Er3+的4F9/2→4I15/2的跃迁。Yb3+具有较大的吸收截面,而Yb3+的2F5/2能级与Er3+的4I11/2能级十分接近,因此在980 nm激发条件下存在着Yb3+-Er3+间的能量传递过程。处于4I11/2上的电子可以再次吸收一个近红外光子,从而跃迁到4F7/2能级。最后通过多声子弛豫和交叉弛豫过程实现4F9/2的布居数累积,实现荧光上转换过程(见图7)。
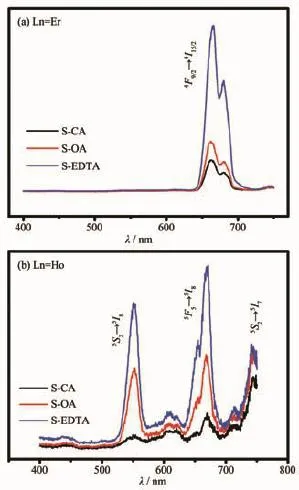
图6 不同添加剂下Gd0.78Yb0.2Ln0.02O3粉末的上转换荧光谱Fig.6 Upconversion Spectra of Gd0.78Yb0.2Ln0.02O3powders derived from different additives
同样地,在980 nm激光激发下,Gd2O3∶Yb3+,Ho3+呈现肉眼可见的荧光。图6(b)中呈现的主要荧光峰位为550,670,742 nm。3个荧光峰位分别对应于Ho3+的5S2→5I8,5F5→5I8,5S2→5I7跃迁。而由于Yb3+的2F5/2能级与Ho3+的5I6能级并不完全匹配,所以通过声子参与的非共振能量传递将能量传递至Ho3+的5I6能级,此能级上的电子通过多声子弛豫、激发态吸收上转换等过程(见图7),最终实现了上述的能量上转换过程。

图7 Yb3+与Er3+,Ho3+之间的能量转移示意图Fig.7 Schematic illustration of energy transfer between Yb3+and Er3+/Ho3+
根据2种稀土掺杂Gd2O3材料的发射光谱可以发现:当样品颗粒变大时,其上转换发光强度均有显著的增强。造成这种现象的原因可能有3个:一是因为样品稀土掺杂Gd2O3颗粒增大时其晶体的非对称性增加[26]。对称性结构下的f-f跃迁是禁阻的,而非对称的结构使得原有f-f跃迁禁阻被部分允许,从而增大了其最终发生辐射跃迁的概率。这种辐射跃迁的概率的增大并不是随着颗粒的增大而线性增大,且对于不同的掺杂对这种效应产生的放大影响亦有不同,但对于我们这个体系,上转换荧光的增强与体系尺寸成正相关。二则是由表面的猝灭中心所导致。一般来说,颗粒的发光中心是由颗粒表面和颗粒内部的发光离子组成,相比于内部的发光中心,表面的发光中心更容易受到表面缺陷引起猝灭的影响而产生荧光衰减[27]。颗粒越小,其比表面则越大,则表面缺陷越多,更容易引发荧光猝灭。对于以上3个样品来说,比表面的大小顺序为:SCA>S-OA>S-EDTA,而从荧光强度来说,S-EDTA>SOA>S-CA,总体上来说符合实验现象。最后一个原因则是从红外谱看出,颗粒表面挂接各种基团,并且从吸收峰强度来看,S-CA>S-OA>S-EDTA,根据Miyakawa-Dexter理论[28],基团的高能振动会增加颗粒的多声子弛豫行为,尤其是那些挂接在Ln3+(Ln= Er3+,Ho3+)离子表面的基团会严重减弱这些激活子的发光强度,因而基团振动吸收峰越大,对荧光产生的负面影响也越大,因此,同样会导致S-EDTA> S-OA>S-CA。
2.4 磁性测量
不同添加剂下的Gd2O3样品的磁化强度M随着所加磁场H(-20~20 kOe)的关系图如图8所示。在不同的添加剂调制下,样品均表现出明显的顺磁性质。在Gd2O3中Gd3+的顺磁性来自于其原子4f层的7个未成对电子,这7个未成对电子受到最外层5s25p6壳层电子的屏蔽,使其不受晶格的晶体场影响。因此,与Gd3+关联的磁矩是局域化的并且是没有相互作用,从而引发了顺磁性[29]。其中,添加剂为EDTA的样品(S-EDTA)表现出最强的顺磁性,S-CA的顺磁性次之,而S-OA的样品顺磁性最弱,具体的质量剩余磁化率分别为1.20×10-4,1.14×10-4,1.01× 10-4emu·g-1·Oe-1,在20 kOe的磁化强度约为2.4,2.28,2.02 emu·g-1,这与之前文献报道的常用生物分离技术的磁化强度相近[30]。造成以上不同剩余磁化率有3个可能的原因:其一是颗粒外面挂接的一些基团分子影响,颗粒表面挂接的一些基团分子如羧基和羟基等可以减弱颗粒的磁性[31],从红外光谱可看出,基团分子C=O和-OH的吸收强度最小的是S-EDTA,因此对磁性的影响最小,其剩余磁化率表现为最大,而S-CA和S-OA的剩余磁化率大小则与红外谱规律相反,可能是不以基团分子的振动吸收强度为主要原因,可以用余下2个原因来解释。第二个因素可能是因为在加入不同的添加剂时其晶格结构会产生变化[32]。由上文可知,3种添加剂作用下的晶格常数分别为:1.082 25 nm(S-CA),1.081 14 nm(S-OA),1.083 20 nm(S-EDTA)。而晶格常数变大意味着原子间距变大,这抑制了Gd原子中4f电子轨道的有效重叠,这种抑制效应使得样品铁磁性减弱,但促使顺磁性增强[31]。最后则可能是与尺寸和形貌有关,不同尺寸和形貌的Gd2O3中的Gd3+分布情况会有所不同,从而导致磁性的变化[33],但目前没有关于这方面的理论报道,需要后续的理论和实验工作支持。

图8 不同添加剂下Gd2O3粉末的室温M-H关系图Fig.8 M-H relationship of Gd2O3powders derived from different additives at room temperature
3 结论
本文通过采取不同添加剂可控合成不同形貌的前驱物Gd(OH)3并通过退火热处理得到Gd2O3及稀土掺杂样品,并且表现出不同的光学和磁学特性。从荧光谱和M-H关系图结果得到,EDTA参与所制得的稀土掺杂Gd2O3样品的上转换发光强度和顺磁磁化率均为最高,这种特性使得EDTA为添加剂的稀土掺杂样品在生物体光学/磁学双探针研究方向具有广大的应用前景。不过EDTA作用下所得Gd2O3产物,其颗粒尺寸较大,难以适用于生物探测领域,因此下一步的工作应该在保持S-EDTA光磁特性最佳的情况下减小其尺寸。本文实验思路为在水热法中采用不同添加剂控制最终产物的形貌以调制稀土氧化物形貌这一领域提供一种参考。
[1]Bridot J,Faure A,Laurent S,et al.J.Am.Chem.Soc.,2007, 129:5076-5084
[2]Lee B,Lee K S,Lee J H,et al.Dalton Trans.,2009,14:2490 -2495
[3]Hong M,Kwo J,Kortan A R,et al.Science,1999,283:1897-1900
[4]Leskel M,Kukli K,Ritala M.J.A lloys Compd.,2006,418: 27-34
[5]Jia G,Liu K,Zheng Y,et al.J.Phys.Chem.C,2009,113: 6050-6055
[6]Bazzi R,Flores-Gonzalez M A,Louis C,et al.J.Colloid Interface Sci.,2004,273:191-197
[7]Bazzi R,Flore-Gonzalez M A,Louis C,et al.J.Lumin., 2003,102-103:445-450
[8]FAN Le-Qing(范乐庆),LI Zhao-Lei(李兆磊),HUANG Yun-Fang(黄昀昉),et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31(1):147-152
[9]Louis C,Bazzi R,Flores-Gonzalez M A,et al.J.Solid State Chem.,2003,172:335-341
[10]Singh S,Kumar K,Rai S.Mater.Sci.Eng.B,2010,166: 180-184
[11]Singh S,Kumar K,Rai S.Appl.Phys.B,2009,94:165-173
[12]Silver J,Martinez-Rubio M L,Ireland T G,et al.J.Phys. Chem.B,2001,105:948-953
[13]Zeng J,Su J,Li Z,et al.Adv.Mater.,2005,17:2119-2123
[14]Yang J,Li C,Cheng Z,et al.J.Phys.Chem.C,2007,111: 18148-18154
[15]Zhou L,Gu Z,Liu X,et al.J.Mater.Chem.,2012,22:966-974
[16]Xing G,Guo Q,Liu Q,et al.Ceram.Int.,2014,40:6569-6577
[17]Zeng S,Tsang M,Chan C,et al.Biomaterials,2012,33:9232 -9238
[18]Guo L,Wang Y,Wang Y,et al.CrystEngComm,2012,14: 3131-3141
[19]Zheng H,Chen B,Yu H,et al.J.Colloid Interface Sci.,2014, 420:27-34
[20]Wang L,He D,Feng S,et al.Sci.Rep.,2014,4:6139
[21]Wild J,Rath J,Meijerink A,et al.Sol.Energ.Mat.Sol.C, 2010,94:2395-2398
[22]Deng K,Gong T,Hu L,et al.Opt.Express,2011,19:1749-1754
[23]Jia G,You H,Liu K,et al.Langmuir,2010,26:5122-5128
[24]Jia G,Yang M,Song Y,et al.Cryst.Growth Des.,2009,9: 301-307
[25]Thirumalai J,Chandramohan R,Valanarasu S,et al.J.Mater. Sci.,2009,44:3889-3899
[26]Ayyub P,Palkar V R,Chattopadhyay S,et al.Phys.Rev.B, 1995,51:6135-6138
[27]Boyer J,Manseau M,Murray J,et al.Langmuir,2009,26: 1157-1164
[28]Miyakawa T,Dexter D L.Phys.Rev.B,1970,1:2961-2969
[29]Wong H,Chan H,Hao J.Appl.Phys.Lett.,2009,95:22512
[30]Yang H,Zhang S,Chen X,et al.Anal.Chem.,2004,76: 1316-1321
[31]Yang L,Zhang Y,Li J,et al.Nanoscale,2012,2:2805-2810
[32]Jiang T,Qin W,Di W,et al.CrystEngComm,2012,14:2302-2307
[33]Chen Z,Liu Z,Liu Y,et al.J.Fluorine Chem.,2012,144: 157-164
Synthesis and Optical/M agnetic Properties of Lanthanides Doped Gd2O3w ith Different M orphologies
XU De-Kang LIU Chu-Feng YAN Jia-Wei OUYANG Hong-Qun ZHANG Yue-Li*
(School of Physics and Engineering,Sun Yat-sen University,Guangzhou 510275,China)
Gd2O3samples(S-CA,S-OA,S-EDTA)with different morphologies are obtained via thermal treatment from the precursors Gd(OH)3,which are hydrothermally prepared using different additives:Citrate acid(CA),Oleic Acid(OA)and Ethylenediaminetetraacetic acid(EDTA).XRD patterns show that the structure of the as-prepared samples are typical cubic phase with Ia3 space group and slight differences in lattice constants:1.082 25 nm(S-CA), 1.081 14 nm(S-OA),1.083 20 nm(S-EDTA).SEM images show that the average particle sizes with different additives are 70 nm(S-CA),300 nm(S-OA),2 μm(S-EDTA),respectively.IR spectra further prove that all the samples are Gd2O3,and samples derived from different additives are coupled with organic groups which have different vibration absorption intensities.Upconversion(UC)luminescent and magnetic properties of Gd0.78Yb0.20Ln0.02O3(Ln=Er,Ho)were also studied.The results show that morphologies have a great impact on lanthanides doped UC luminescence intensity and paramagnetic susceptibility of Gd2O3samples.Among them,both UC luminescence intensity and paramagnetic susceptibility of rare-earth doped Gd2O3powders derived from EDTA reach the optimum values.
rare-earth oxides;additive;morphology;lum inescence;magnetic properties
TQ133
A
1001-4861(2015)04-0689-07
10.11862/CJIC.2015.076
2014-09-01。收修改稿日期:2014-12-11。
国家自然科学基金(No.61172027);广东省自然科学基金重点(No.1414050000317)资助项目。*
猜你喜欢
稀土信息(2022年1期)2022-02-15
稀土信息(2022年1期)2022-02-15
陶瓷学报(2020年2期)2020-10-27
四川冶金(2019年5期)2019-12-23
铜仁学院学报(2018年6期)2018-07-05
资源节约与环保(2018年1期)2018-02-08
中国资源综合利用(2017年4期)2018-01-22
广州化学(2016年5期)2016-11-15
衡阳师范学院学报(2016年3期)2016-07-10
当代化工研究(2016年7期)2016-03-20
