
四氢原小檗碱类化合物两条全合成路线的探讨*
2015-12-05 07:26:38徐淑华宋文文葛海霞
湖州师范学院学报 2015年10期
李 昌,王 飞,徐淑华,宋文文,葛海霞
(湖州师范学院 生命科学学院,浙江 湖州313000)
0 引言
四氢原小檗碱类化合物(THPBs)是一类广泛存在于中药延胡索(corydalis)、千金藤(stephania)及地不容等植物中的天然活性成分[1].THPBs类化合物具有多种生物活性.在中枢神经系统方面[2~3],THPBs类化合物具有很好的镇痛、抗帕金森症、抗精神分裂症和抗药物成瘾等活性,该活性主要与脑内多巴胺(DA)受体功能相关,开拓了四氢原小檗碱同类物作用于脑内DA 受体的研究领域,把中药THPBs作用融入现代DA 的科学范畴中;在心血管系统方面[1],THPBs类化合物具有较强的降血压、增加冠脉流量、保护心肌及抗心律失常等作用,其机制主要与拮抗α受体和β受体、抑制相关离子通道等相关.此外还有一定的抗炎、抗肿瘤、抗菌和抗疟等活性[4].构效关系研究表明[5~6],THPBs的活性与A 环和D 环上的取代基不同密切相关,A 环和D 环上的取代基不同使THPBs的扭曲度不同,进而使二面角的角度不同,引起活性的差异.因此,制备在A 环和D 环有多种不同的取代基的THPBs衍生物对开发该类化合物具有重要意义.
THPBs的制备方法主要分为两大类:一类是全合成方法;另一类是以天然产物盐酸小檗碱等为原料的半合成方法.半合成方法相对步骤较少,操作更简便,但制备化合物的种类有限.全合成方法可在其结构的不同位置引入不同取代基获得取代基多样的THPBs类化合物.
目前,四氢原小檗碱类衍生物最常用的两条全合成路线如图1和图2所示.本文主要对这两条全合成路线每一步反应的特点以及各自优缺点进行探索性的评价.
1 以取代苯甲醛为起始原料合成THPBs
如图1所示,该路线[7]是以胡椒乙胺或高藜芦胺等和多种取代苯甲醛作为起始原料,通过缩合反应、还原反应、环合反应、碱化脱铜反应、还原反应五步反应制备THPBs类化合物.
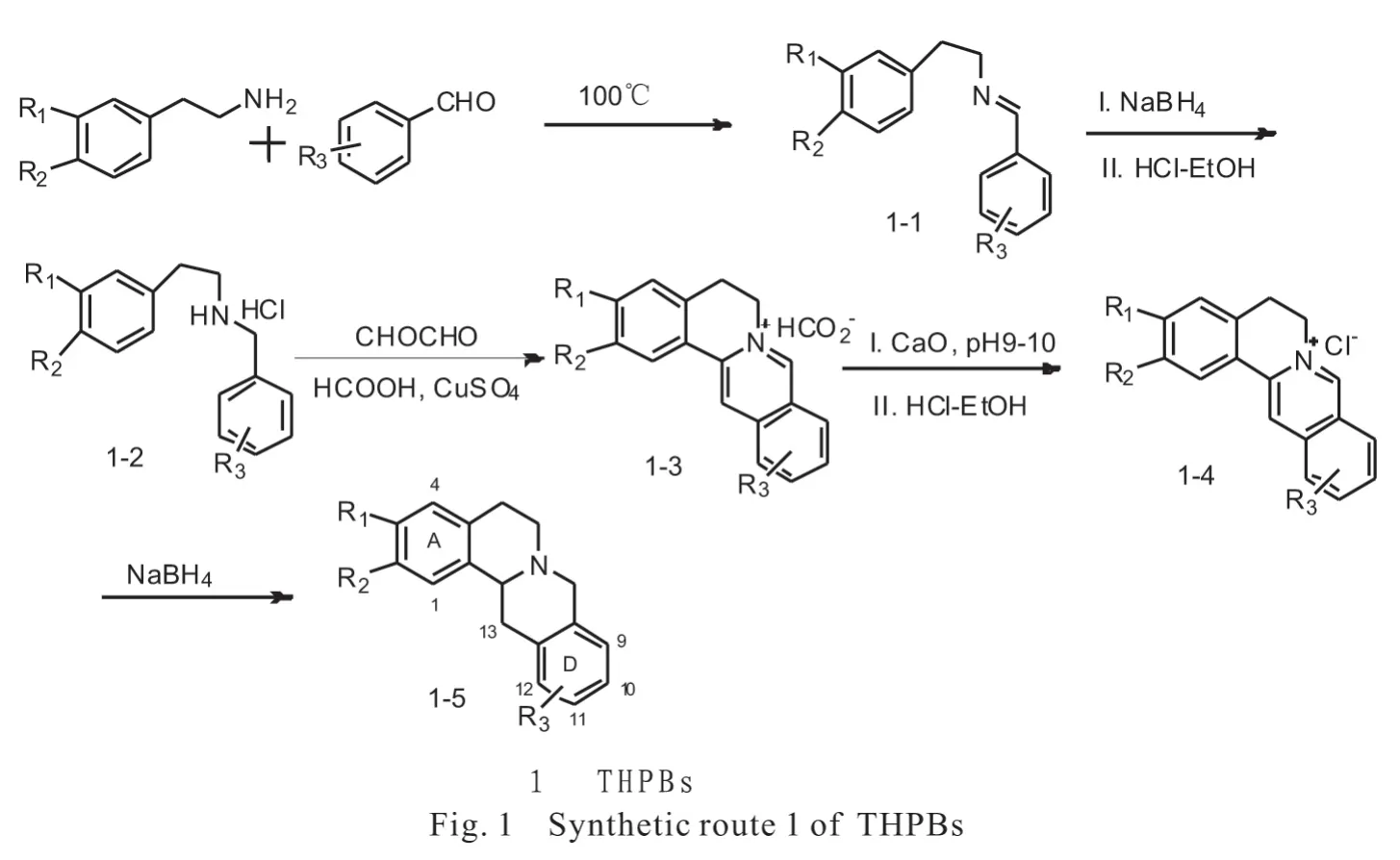
在该合成路线中,第一步反应是在100 ℃加热脱水缩合至TLC 监测反应完全为止,生成的产物1-1不需纯化处理即可直接进行下一步反应.第二步反应是将上步产物溶解于乙醇中,加入过量3~5倍摩尔数的NaBH4反应至TLC监测原料消失为止,经萃取纯化后,再用盐酸-乙醇(5∶95)酸化成盐,以上两步的总收率约为78%.第三步环合反应是整个反应过程中最难、最关键的一步,直接决定最终产物的收率.以无水甲酸为溶剂,无水硫酸铜作为脱水剂,在100 ℃条件下反应,加入40%乙二醛溶液提供环合反应所需要的“二碳单位”,且在反应过程中分多次加入浓盐酸作为催化剂.在进行多次该步反应时发现,硫酸铜和浓盐酸的用量、反应温度、投料时间和顺序等多个因素对该步产物的收率都有很大影响,其中硫酸铜用量影响最大,用量过多会使其反应的后处理困难,用量过少则又无法除去溶剂及反应中产生的水,使产率大大降低.第四步碱化脱铜反应是将上步反应产物用甲醇和水溶解,再用氧化钙调pH 至9~10.直接加氧化钙粉末调pH 值时很难准确控制其所需的量,用量过多会使后处理抽滤的时间大大延长,而且会吸附一定量的产物,造成损失,进而直接影响最后的收率;用量过少,则会导致产物脱铜不完全,无法完全除去铜离子而导致产物颜色加深,增加纯化的困难.此外调好pH 值后搅拌的时间也不能过长,否则料液很粘稠,给后处理造成很大的麻烦.总之,该步反应的操作较繁琐,需要不断摸索改进方法.碱化脱酮处理后的产物还需要用盐酸-乙醇(5∶95)进一步酸化成盐,再经硅胶柱层析分离纯化得到目标物1-4.以上两步反应的总收率约为25%~45%.第五步还原反应方法同第二步,采用3~5倍摩尔量的NaBH4使反应完全,通过重结晶方法得到高纯度的目标物1-5,该步收率约为80%.
采用该路线一,本文尝试以多种不同取代基的苯甲醛与胡椒乙胺或高藜芦胺等作为起始原料,制备了近20个THPBs类化合物,化合物的总收率为15%~30%.在制备不同的THPBs类化合物过程中发现:①当A 环没有任何取代基时,即起始原料用苯乙胺与取代的苯甲醛进行反应,环化反应很难进行,产率很低或基本不发生反应;②当A 环的2,3位有甲氧基或二氧亚甲基等基团时,若D 环同时存在10-位给电子基,如甲氧基或羟基时,环化反应收率较高,可达45%左右;若D 环10-位没有给电子基,而其它位如9-位或11-位等有给电子基时,产率非常低,且无法分离到产物;若D 环没有取代基或有吸电子基,如氯、硝基时,不能发生环化反应.这些现象与其反应机理密切相关.
2 以取代苯乙酸为起始原料合成THPBs
如图2所示,该路线[8]是以胡椒乙胺或高藜芦胺等和多种取代的苯乙酸作为起始原料,通过缩合反应、脱水环合B环反应、还原反应、脱水环合C环四步反应获得目标物.

在该合成路线中,如果采用的起始原料苯乙酸结构中有羟基或胺基取代基时,需要将其进行保护,如羟基可采用苄基保护、胺基可采用BOC保护,再进行以下步骤反应.第一步缩合反应根据文献[8]的方法将起始原料胺与取代苯乙酸在170~180 ℃加热反应3~4h获得缩合产物2-1.但用胡椒乙胺与邻羟基苯乙酸缩合反应时,发现该方法反应不完全,收率很低.通过查阅文献[9],将反应方法改为以EDCI/HOBT为缩合剂,室温反应过夜,即可反应完全得到缩合产物2-1,收率约为80%.第二步脱水环合B环反应必须在无水环境中进行,加入过量的三氯氧磷在110 ℃下反应4h,可得到收率约为70%的产物2-2.第三步还原反应采用甲醇为反应溶剂,过量的硼氢化钠为还原剂,即可得到反应产物2-3,但在后处理的萃取时易产生严重的乳化现象,收率约为70%.第四步脱水环合C 环反应.根据文献[8]的方法,用98%甲酸和37%甲醛在100 ℃下反应3h,后处理应得到目标产物2-4,但以邻苄氧基苯乙酸或邻甲氧基苯乙酸为原料进行该步反应时,经波谱鉴定该步得到的产物并非目标产物2-4,而是上步目标物2-3的N 甲基化产物.通过查阅文献[10],将此步反应的方法改为:往上步产物2-3中加入新制备的甲乙酐,室温搅拌反应过夜,经萃取等后处理即可得到N 甲酰化产物2-5,再将2-5经脱水环合C环反应和还原反应最终得到目标物2-4,通过硅胶柱层析分离纯化得到纯品目标物.但这三步的综合收率只有15%左右,特别是最后一步脱水环合C环反应不完全,导致收率很低.
采用该合成路线二,本文尝试以胡椒乙胺分别与邻甲氧基苯乙酸和邻苄氧基苯乙酸作为起始原料,制备了两个THPBs类化合物,其总收率只有5%左右,而且纯化较难.
据文献报道[11~13],该合成路线二的第四步脱水环合C 环反应中甲酸和甲醛加热反应条件下易环合,且收率可达80%.但本文尝试以邻甲氧基苯乙酸和邻苄氧基苯乙酸为起始原料在该步无法得到目标物,这是否与苯乙酸结构上取代基所在的位置有关系,有待进一步研究.
3 结语
在四氢原小檗碱类化合物的两条常用的全合成路线中,路线一与路线二相比,反应步骤更短,收率更高,总收率可达15%~30%,而且不同取代基的苯甲醛种类多,价格相对便宜,更适合制备结构多样的四氢原小檗碱类化合物;路线二反应步骤较长,收率较低,而且不同取代基的苯乙酸种类有限,可作为路线一的替补方法合成更多样的THPBs类化合物.
[1]金国章.中药延胡索研究中的新发现[M].上海:上海科技出版社,2001.
[2]Mo J,Guo Y,Yang Y S,et al.Recent developments in studies of l-stepholidine and its analogs:chemistry,pharmacology and clinical implications[J].Curr Med Chem,2007,14:2 996-3 002.
[3]Chu H Y,Jin G Z,Friedman E,et al.Recent development in studies of tetrahydroproto berberines:mechanism in antinociception and drug Addiction[J].Cell Mol Neurobiol,2008,28:491-499.
[4]Emidio V L D,Ivana M M F,Diego N G.The Alkaloids:Chemistry and Biology[M].New York:Academic Press,2005.
[5]Xuan J C,Lin G D,Jin G Z,et al.Relevance of stereo and quantum chemistry of four tetrahydroprotoberberines to their effects on dopamine receptors[J].Acta Pharmacol Sin,1988(9):197-205.
[6]吴安,陈洁,金国章,等.四氢原小檗碱同类物的晶体结构对D1和D2多巴胺受体亚型的作用关系[J].自然科学进展,1991(2):147-152.
[7]Yang P,Song D Q,Li Y H,et al.Synthesis and structure-activity relationships of berberine analogues as a novel class of low-density-lipoprotein receptor up-regulators[J].Bioorg Med Chem Lett,2008,18:4 675-4 677.
[8]Kametani T,Shibuya S,Hirata S,et al.Studies on the syntheses of heterocyclic compounds.D.novel cleavage of tetrahydroprotoberberines with trifluoroacetic anhydride[J].Chem Pharm Bull,1972,20(12):2 570-2 574.
[9]Fanrong M,Stephanie L C,David J R,et al.Design,synthesis,and biological evaluation of a series of lavendustin a analogues that inhibit EGFR and syk tyrosine kinases,as well as tubulin polymerization[J].J Med Chem,2001,44:441-452.
[10]Hanaoka M,Kobayashi N,Shimada K I,et al.Chemical transformation of protoberberines.Part 10,a novel synthesis of sanguilutine and dihydrosanguilutine,fully aromatized 2,3,7,8,10-pentaoxygenated benzo[c]phenanthridine alkaloids[J].J Chem Soc Perkin Trans I,1978(6):677-681.
[11]Ishiwata S,Itakura K.Syntheses of aminoisoquinolines and related compounds.V.The direaction of the mannich reaction on the aminotetrahydroisoquinolines to the aminoprotoberberines[J].Chem Pharm Bull,1970,18(5):896-900.
[12]Zhai H M,Miller J,Sammis G.First enantioselective syntheses of the dopamine D1and D2receptor modulators,(+)-and(-)-govadine[J].Bioorg Med Chem Lett,2012,22:1 557-1 559.
[13]Parraga J,Cabedo N,Andujar S,et al.2,3,9-and 2,3,11-Trisubstituted tetrahydroprotoberberines as D2dopaminergic ligands[J].Eur J Med Chem,2013,68:150-166.
猜你喜欢
西南农业学报(2020年8期)2020-12-10 01:50:02
中成药(2018年12期)2018-12-29 12:26:08
中成药(2018年12期)2018-12-29 12:25:18
中成药(2018年3期)2018-05-07 13:34:18
临床肝胆病杂志(2017年3期)2017-03-07 06:44:51
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:36:55
合成化学(2015年1期)2016-01-17 08:55:47
应用化工(2014年12期)2014-08-16 13:10:46
天然产物研究与开发(2014年7期)2014-04-27 14:16:08
山西大学学报(自然科学版)(2013年1期)2013-10-23 09:20:20
