
乳腺癌、甲状腺癌和黑色素瘤全基因组关联分析计量和热点研究
2015-11-28 02:10许春伟
承德医学院学报 2015年3期
王 斌,许春伟
(1.唐县人民医院病理科,河北唐县 723502;2.中国人民解放军北京军区总医院病理科)
乳腺癌、甲状腺癌和黑色素瘤全基因组关联分析计量和热点研究
王 斌1,许春伟2△
(1.唐县人民医院病理科,河北唐县 723502;2.中国人民解放军北京军区总医院病理科)
全基因组关联研究;GWAS;文献研究
全基因组关联研究(GWAS)是利用高通量基因芯片技术,对人类全基因组范围内常见遗传变异-单核苷酸多态性(SNP)和拷贝数变异(CNV)进行总体关联分析的研究方法[1]。国际人类基因组测序和单体型图谱工程的完成,以及经济高效的高通量基因分型技术的开展,使全基因组范围内筛检与疾病相关的序列变异成为可能,通过比较全基因组范围内所有变异的等位基因频率,从中发现与疾病相关联的序列变异[2-3]。2007年,Duggan等[4]和Murabito等[5]利用Illumina及Affymetrix的基因芯片对前列腺癌进行了GWAS,此后,越来越多肿瘤方面的GWAS研究结果陆续发表。本研究使用最新的GWAS数据库之一—NHGRI,分析挖掘乳腺癌、甲状腺癌和黑色素瘤文献特征数据,以期为国内甲状腺、乳腺及皮肤肿瘤GWAS提供参考。
1 材料与方法
研究资料来源于NHGRI。登入页面http://www.genome. gov/page.cfm?pageid=26525384#searchForm,在页面下Disease/ Trait一栏中输入“Breast cancer”,点击Search后再点击“Download Spreadsheet of Search Result”保存“MyGWASSearch. xls”,并改名为“Breast cancer.xls”,并用同样的步骤获得“Thyroid cancer.xls”和“Melanoma.xls”。
2 结果
2.1 文献基本特征 数据库原记录包含34篇论文[6-38](乳腺癌24篇、甲状腺癌2篇、黑色瘤7篇),涉及114个SNP(乳腺癌88个,甲状腺癌5个,黑色素瘤21个)。论文分别发表在Nat Genet(IF:35.209/2012)14篇,Nature(IF:38.597/2012)1篇,Breast Cancer Res (IF:5.872/2012)2篇,Breast Cancer Res Treat (IF:4.469/ 2012)2篇,Hum Genet(IF:4.633/2012)4篇,Hum Mol Genet(IF:7.692/ 2012)3篇,J Hum Genet(IF:2.365/2012)1篇,J Natl Cancer Inst(IF:14.336/2012)1篇,PLoS Genet (IF:8.517/2012)3篇,PLoS One(IF:3.73/2012)1篇,Proc Natl Acad Sci U S A(IF:9.737/2012)1篇。
2.2 第一(筛选)阶段样本量 34篇乳腺、甲状腺及皮肤肿瘤GWAS研究文献第一阶段病例组样本量30-27758例不等,对照组例数30-37196例。按病例数/对照数的研究文献分布见图1。5.88%文献没有明确报告对照组样本量。

图1 乳腺、甲状腺及皮肤肿瘤GWAS文献第一阶样本量分布
2.3 第二(验证)阶段样本量 第二阶段病例组样本量203-26646例,没有报告第二阶段病例数据的文献占11.76%;对照组例数263-55667例,2.94%的文献没有报告对照组样本量。按病例数/对照数的研究文献分布见图2。
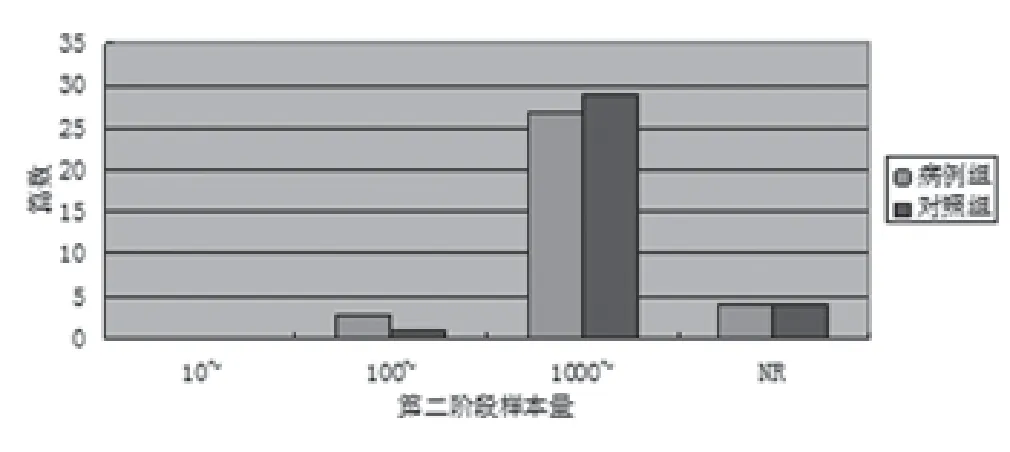
图2 乳腺、甲状腺及皮肤肿瘤GWAS文献第二阶段样本量分布
2.4 染色体区域相关SNP(NCBI rs登记号) 所定位的染色体区域分布大致与染色体长度近似,其中10号和16号染色体明显多于邻近的染色体(图3)。
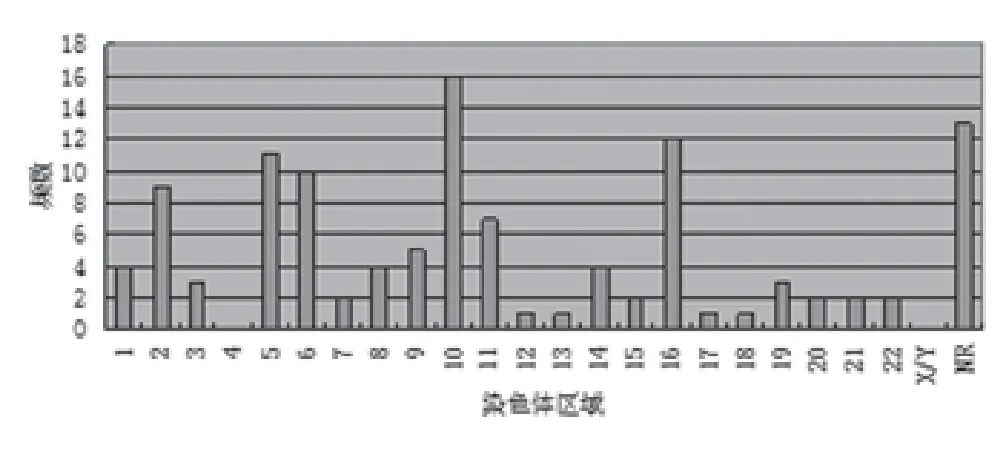
图3 乳腺、甲状腺及皮肤肿瘤GWAS文献中有明确rs(NCBI)登记号SNP所在染色体区域
2.5 对照组相关风险等位基因频率 对照组中与最强SNP相关风险等位基因频率在乳腺、甲状腺及皮肤肿瘤GWAS文献中的分布,未发现稀有变异(即在对照人群中的发生频率<1%)的乳腺、甲状腺及皮肤肿瘤GWAS文献(图4)。
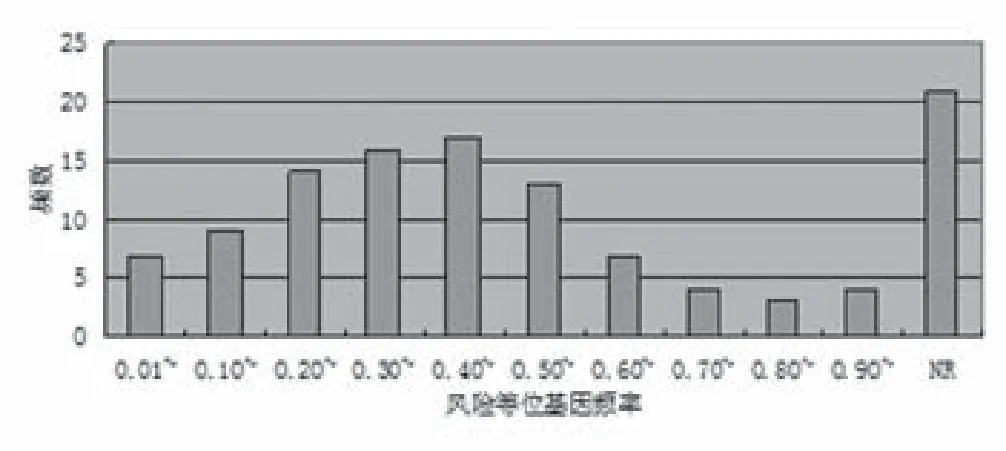
图4 对照组中与最强SNP相关风险等位基因频率在乳腺、甲状腺及皮肤肿瘤GWAS文献中的分布
2.6 P值文献中最强SNP 相关风险等位基因P值选择<1× 10-6为录入标准,最小P值2×10-76。P值分布见图5:

图5 P值分布
2.7 优势比(OR)或β相关系数 文献中最强SNP相关风险等位基因优势比(或β相关系数)为1.04-8.40(图6)。
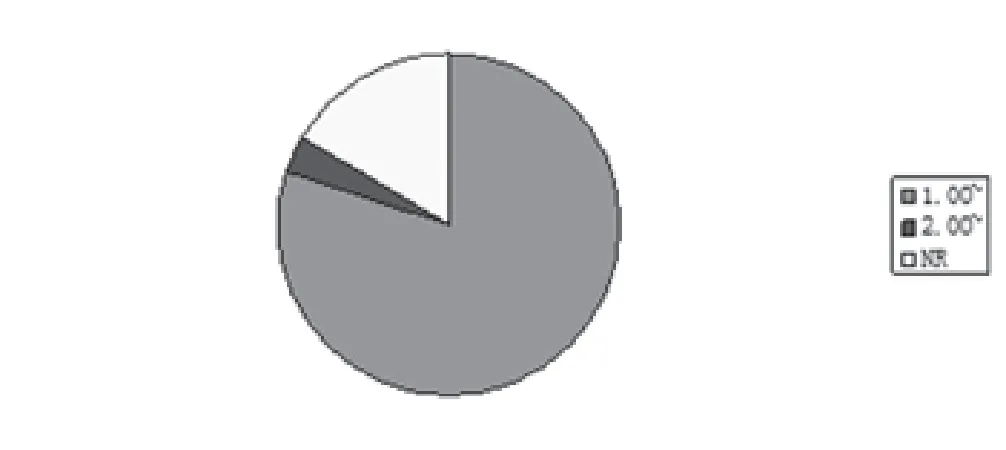
图6 最强SNP相关风险等位基因优势比(或β相关系数)分布
2.8 实验平台及检测SNP数量 超过一半(64.70%)的乳腺、甲状腺及皮肤肿瘤GWAS独立使用了Illumina研究平台,近三分之一(32.35%)使用Affimetrix芯片系统,2.94%使用Perlegen芯片系统。检测SNP数量70897-5480804个,14.71%的研究用试验平台的SNP数量在100万-1000万之间(图7-8)。
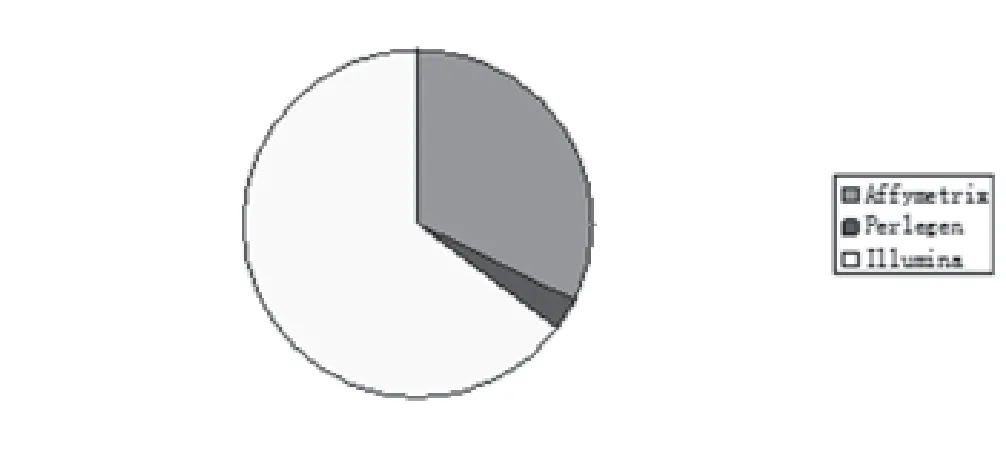
图7 乳腺、甲状腺及皮肤肿瘤GWAS文献实验的试验平台分布
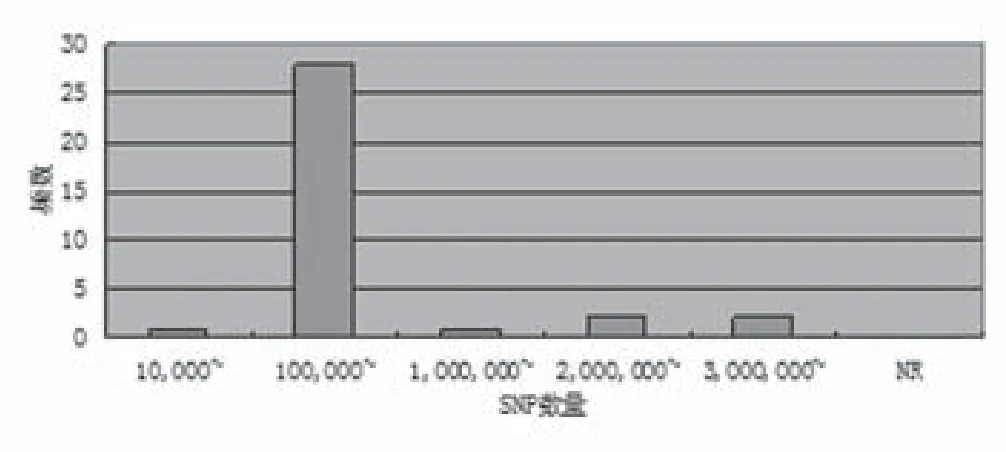
图8 乳腺、甲状腺及皮肤肿瘤GWAS文献使用的试验平台检测SNP数量
3 讨论
3.1 概述 目前,乳腺、甲状腺及皮肤肿瘤GWAS方面的报道主要在甲状腺癌、乳腺癌和黑色素瘤领域。乳腺癌GWAS方面,Easton等[26]研究发现,10q26.1区域(FGFR2基因)rs2981582、16q12.1区域(TNRC9/LOC643714基因)rs3803662、5q11.2区域(MAP3K1基因)rs889312、11p15.5区域(LSP1基因)rs3817198、5q11.2(基因间隔区)rs13281615和5p12(基因间隔区)rs981782位点多态性与乳腺癌易感性相关联;Fletcher等[18]研究发现,9q31.2区域(KLF4/RAD23B/ ACTL7A基因)rs865686位点多态性与乳腺癌易感性相关,并验证了Easton等基因多态性位点。甲状腺癌GWAS方面,Gudmundsson等[7]研究发现,9q22.3区域(FOXE1基因)rs965513和14q13.3区域(NKX2-1基因)rs944289位点多态性与低浓度的甲状腺激素(TSH)显著相关,其中rs965513与低浓度的甲状腺素(T4)和高浓度的甲状腺(T3)相关;三年后,Gudmundsson等又报道了2q35区域(DIRC3基因)rs966423、8p12区域(NRG1基因)rs2439302和14q13区域(MBIP基因)rs116909374位点多态性与甲状腺癌相关。黑色素瘤GWAS方面,Brown等[38]研究发现20q11.2区域(CDC91L1基因)rs910873位点多态性与黑色素瘤相关;随后,Barrett等报道了2q33区域(CASP8基因)rs13016963、11q22.3区域(ATM基因)rs1801516、21q22.3区域(MX2基因)rs45430、5p15.3区域(TERT/CLPTM1L基因)rs401681、5p13.2区域(SLC45A2基因)rs35390、9p21区域(CDKN2A/MTAP基因)rs7023329、11q14.3区域(TYR基因)rs1393350、16q24区域(MC1R基因)rs258322、6q23.2区域(ASIP基因)rs2284378和22q13.1区域(PLA2G6基因)rs6001027位点多态性与黑色素瘤相关。
3.2 NHGRI数据库分析 通过对NHGRI数据库中乳腺、甲状腺及皮肤肿瘤中八项数据进行不完全统计发现,乳腺、甲状腺及皮肤肿瘤有如下特点:⑴在文献基本特征方面:乳腺、甲状腺及皮肤肿瘤在Nat Genet发表的文献最多,占41.18%,其次是Hum Genet(11.76%)和Hum Mol Genet、PLoS Genet(8.82%),再次是Breast Cancer Res Treat(5.88%)。因此推测,Nat Genet以其影响力将是优质乳腺、甲状腺及皮肤肿瘤GWAS的主要阵地。⑵甲状腺、乳腺及皮肤肿瘤GWAS第一、二阶段的病例组样本量,一般从几百例到几万例不等,且以千例为主,最大样本量均不超过30000例;对照组样本量也从几百例到几万例不等,但以千例以上为主,最大样本量均超过60000例。⑶染色体区域相关SNP方面:以10号和16号染色体区域SNP居多,其中10号染色体区域SNP在消化系统肿瘤中也占一定数量,16号染色体区域SNP在其它肿瘤中比较少见。⑷对照组相关风险等位基因频率方面:乳腺、甲状腺及皮肤肿瘤GWAS中风险等位基因以Risk Allele Frequency=0.40左右最多,其次是Risk Allele Frequency=0.30,但未发现稀有变异。⑸P值文献中最强SNP方面:乳腺、甲状腺及皮肤肿瘤P值以10-6和10-10为主,其中最强P值2×10-76,是目前为止所有肿瘤GWAS中P值最高的。⑹优势比(OR)或β相关系数方面:乳腺、甲状腺及皮肤肿瘤OR以1.00左右的弱相关占多数,2.00以上的强相关所占比例较少。⑺实验平台及检测SNP数量方面:乳腺、甲状腺及皮肤肿瘤实验平台方面Illumina研究平台占大多数,其次是Affymetrix芯片系统和Perlegen研究平台,未见Invader研究平台的报道。检测SNP数量从70897个到5480804个,近1/6试验平台的SNP数量在100万-1000万之间。
综上所述,肿瘤方面GWAS的最终目标是应用于临床,主要体现在对正常人群进行肿瘤的危险度预测评估和指导肿瘤的个体化治疗。个体化治疗推崇在肿瘤遗传谱指导下的个体化医学,为肿瘤发生和治疗疗效提供早期预测,以促进肿瘤的预防和治疗。乳腺、甲状腺及皮肤肿瘤GWAS经过7年的发展,已在乳腺癌、甲状腺癌和黑色素瘤遗传易感性研究方面取得了很大成功,为个体化治疗打下了基础。本文通过运用文献计量学方法客观分析了NHGRI的文献基本特征、第一、二阶段样本量、染色体区域相关SNP、对照组相关风险等位基因频率、P值文献中最强SNP、优势比(OR)或β相关系数和实验平台及检测SNP数量八项数据,找到了乳腺、甲状腺及皮肤肿瘤GWAS的一些共性特征,可为乳腺、甲状腺及皮肤肿瘤GWAS的研究现状和发展趋势奠定基础,并可为国内乳腺、甲状腺及皮肤肿瘤GWAS提供参考。
[1]张学军.全基因组关联分析对银屑病遗传学研究的启示[J].浙江大学学报(医学版),2009,38(4):333-337.
[2]严卫丽.复杂疾病全基因组关联研究进展—研究设计和遗传标记[J].遗传,2008,30(4):400-406.
[3]严卫丽.复杂疾病全基因组关联研究进展—遗传统计分析[J].遗传,2008,30(5):543-549.
[4]Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP[J]. J Natl Cancer Inst,2007, 99(24): 1836-1844.
[5]Murabito JM, Rosenberg CL, Finger D, et al. A genome-wide association study of breast and prostate cancer in the NHLBI’s Framingham Heart Study[J]. BMC Med Genet, 2007, 8 Suppl 1:S6.
[6]Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk[J]. Nat Genet, 2012,44(3): 319-322.
[7]Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations[J]. Nat Genet, 2009, 41(4): 460-464.
[8]Kim HC, Lee JY, Sung H, et al. A genome-wide association study identif ies a breast cancer risk variant in ERBB4 at 2q34: results from the Seoul Breast Cancer Study[J]. Breast Cancer Res, 2012,14(2): R56.
[9]Li J, Humphreys K, Darabi H, et al. A genome-wide association scan on estrogen receptor-negative breast cancer[J]. Breast Cancer Res, 2010, 12(6): R93.
[10]Li J, Humphreys K, Heikkinen T, et al. A combined analysis of genome-wide association studies in breast cancer[J]. Breast Cancer Res Treat, 2011, 126(3): 717-727.
[11]Kibriya MG, Jasmine F, Argos M, et al. A pilot genome-wide association study of early-onset breast cancer[J]. Breast Cancer Res Treat, 2009, 114(3): 463-477.
[12]Rinella ES, Shao Y, Yackowski L, et al. Genetic variants associated with breast cancer risk for Ashkenazi Jewish women with strong family histories but no identifiable BRCA1/2 mutation[J]. Hum Genet, 2013, 132(5): 523-536.
[13]Chen F, Chen GK, Stram DO, et al. A genome-wide association study of breast cancer in women of African ancestry[J]. Hum Genet, 2013, 132(1): 39-48.
[14]Sehrawat B, Sridharan M, Ghosh S, et al. Potential novelcandidate polymorphisms identif ied in genome-wide association study for breast cancer susceptibility[J]. Hum Genet, 2011,130(4): 529-537.
[15]Siddiq A, Couch FJ, Chen GK, et al. A meta-analysis of genomewide association studies of breast cancer identifies two novel susceptibility loci at 6q14 and 20q11[J]. Hum Mol Genet, 2012,21(24): 5373-5384.
[16]Cai Q, Long J, Lu W, et al. Genome-wide association study identif ies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium[J]. Hum Mol Genet, 2011,20(24): 4991-4999.
[17]Elgazzar S, Zembutsu H, Takahashi A, et al. A genome-wide association study identif ies a genetic variant in the SIAH2 locus associated with hormonal receptor-positive breast cancer in Japanese[J]. J Hum Genet, 2012, 57(12): 766-771.
[18]Fletcher O, Johnson N, Orr N, et al. Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study[J]. J Natl Cancer Inst, 2011, 103(5): 425-435.
[19]Haiman CA, Chen GK, Vachon CM, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptornegative breast cancer[J]. Nat Genet, 2011, 43(12): 1210-1214.
[20]Antoniou AC, Wang X, Fredericksen ZS, et al. A locus on 19p13 modif ies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population[J]. Nat Genet, 2010, 42(10): 885-892.
[21]Turnbull C, Ahmed S, Morrison J, et al. Genome-wide association study identif ies f ive new breast cancer susceptibility loci[J]. Nat Genet, 2010, 42(6): 504-507.
[22]Thomas G, Jacobs KB, Kraft P, et al. A multistage genome-wide association study in breast cancer identif ies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1)[J]. Nat Genet, 2009, 41(5):579-584.
[23]Zheng W, Long J, Gao YT, et al. Genome-wide association study identif ies a new breast cancer susceptibility locus at 6q25.1[J]. Nat Genet, 2009, 41(3): 324-328.
[24]Hunter DJ, Kraft P, Jacobs KB, et al. A genome-wide association study identif ies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer[J]. Nat Genet, 2007, 39(7):870-874.
[25]Stacey SN, Manolescu A, Sulem P, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer[J]. Nat Genet, 2007, 39(7):865-869.
[26]Easton DF, Pooley KA, Dunning AM, et al. Genome-wide association study identifies novel breast cancer susceptibility loci[J]. Nature, 2007, 447(7148): 1087-1093.
[27]Long J, Cai Q, Sung H, et al. Genome-wide association study in east Asians identif ies novel susceptibility loci for breast cancer[J]. PLoS Genet, 2012, 8(2): e1002532.
[28]Gaudet MM, Kirchhoff T, Green T, et al. Common genetic variants and modification of penetrance of BRCA2-associated breast cancer[J]. PLoS Genet, 2010, 6(10):e1001183.
[29]Long J, Cai Q, Shu XO, et al. Identif ication of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium[J]. PLoS Genet, 2010, 6(6): e1001002.
[30]Song C, Chen GK, Millikan RC, et al. A genome-wide scan for breast cancer risk haplotypes among African American women[J]. PLoS One, 2013, 8(2): e57298.
[31]Gold B, Kirchhoff T, Stefanov S, et al. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33[J]. Proc Natl Acad Sci U S A, 2008, 105(11): 4340-4345.
[32]Teerlink C, Farnham J, Allen-Brady K, et al. A unique genomewide association analysis in extended Utah high-risk pedigrees identifies a novel melanoma risk variant on chromosome arm 10q[J]. Hum Genet, 2012, 131(1): 77-85.
[33]Amos CI, Wang LE, Lee JE, et al. Genome-wide association study identif ies novel loci predisposing to cutaneous melanoma[J]. Hum Mol Genet, 2011, 20(24): 5012-5023.
[34]Iles MM, Law MH, Stacey SN, et al. A variant in FTO shows association with melanoma risk not due to BMI[J]. Nat Genet,2013, 45(4):428-432, 432e1.
[35]Barrett JH, Iles MM, Harland M, et al. Genome-wide association study identifies three new melanoma susceptibility loci[J]. Nat Genet, 2011, 43(11): 1108-1113.
[36]Macgregor S, Montgomery GW, Liu JZ, et al. Genome-wide association study identif ies a new melanoma susceptibility locus at 1q21.3[J]. Nat Genet, 2011, 43(11): 1114-1118.
[37]Bishop DT, Demenais F, Iles MM, et al. Genome-wide association study identifi es three loci associated with melanoma risk[J]. Nat Genet, 2009, 41(8): 920-925.
[38]Brown KM, Macgregor S, Montgomery GW, et al. Common sequence variants on 20q11.22 confer melanoma susceptibility[J]. Nat Genet, 2008, 40(7): 838-840.
R73
C
1004-6879(2015)03-0267-04
2014-06-23)
猜你喜欢
内蒙古统计(2021年4期)2021-12-06
今日农业(2021年11期)2021-08-13
国际放射医学核医学杂志(2021年10期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中国卫生统计(2019年3期)2019-07-10
中成药(2018年7期)2018-08-04
中国卫生标准管理(2015年2期)2016-01-15
中国中西医结合外科杂志(2013年3期)2013-03-11
