
头孢西丁合成工艺的改进
2015-11-28 12:44黄伟平虞正烨何发明何小鹏金立阔
浙江化工 2015年4期
黄伟平,虞正烨,何发明,何小鹏,金立阔
(浙江东邦药业有限公司,浙江临海317016)
头孢西丁合成工艺的改进
黄伟平,虞正烨,何发明,何小鹏,金立阔
(浙江东邦药业有限公司,浙江临海317016)
以7-ACA为起始原料,通过酰胺化、甲氧基化、去乙酰化、甲氨酰化反应,合成得到头孢西丁,其中酰胺化、甲氧基化产物不用结晶提纯,直接进行下步反应,缩短了工业生产周期和节省了原料。本方法制备简便,适于工业化生产。
工艺改进;头孢西丁酸;合成
头孢西丁,化学名为(6R,7S)-3-氨基-甲酰基氧-甲基-7-甲氧基-8-氧代-7-2[(2-吩噻基)乙酰氨基-5-硫-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸,由美国Merk公司研制。头孢西丁是由链霉菌产生的甲氧头孢菌素C经半合成制得的一类新型抗生素,由于抑制细菌细胞壁合成而杀灭细菌,且对细菌产生的内酰胺酶具有很高的抵抗性,因而具有巨大的市场潜力[1-3]。
头孢西丁的合成路线主要有3条:
(1)路线1[4]以头孢霉素C为起始原料,首先对C7位侧链氨基和C3位羧基进行保护,然后氨基接上侧链噻吩乙酰基,最后脱去氨基和羧基保护基得到头孢西丁;该路线起始原料价格较高,不易获得,故生产原料成本高,一直没有满足工业化大生产的需求。
(2)路线2[5]以头孢噻吩酸或其钠盐为起始原料,首先进行甲氧基化反应,然后在低温强碱作用下水解脱去乙酰基,最后C3羟基进行氨甲基化得到目标产物头孢西丁。该路线起始原料易得,路线简单,适合工业化生产。

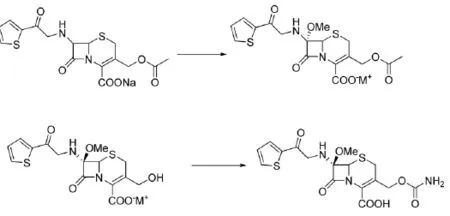
(3)路线3[6]以7-MAC为起始原料,首先进行酰化反应接上侧链噻吩乙酰基,然后在低温强碱作用下水解脱去乙酰基,最后C-3羟基进行氨甲基化得到目标产物头孢西丁。该路线起始原料虽然易得,但价格较贵,原料成本高,没有工业化优势。
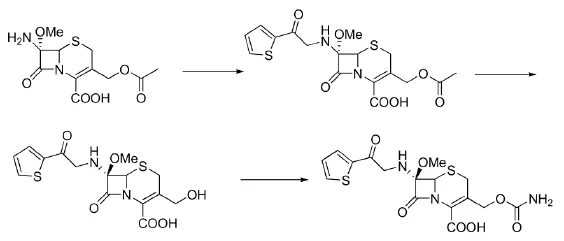
参考路线2,我们以更为便宜易得的7-ACA为起始原料,首先采用改进工艺合成头孢噻吩酸,对于头孢噻吩酸的合成,文献[7]采用在碳酸氢钠水溶液体系反应,由于β-内酰胺类化合物在碱性水溶液易于降解,收率81.7%,经过改进,我们采用三乙胺、二氯甲烷加微量水体系,反应收率提高到95%,而且纯度也由98%提高到98.7%,由于质量的提高,考虑到生产的简单化,头孢噻吩酸不需结晶直接进行甲氧基化,甲氧基化产物也不需要结晶,直接进行下一步酶解;关于去乙酰基化,我们避免了原始路线低温强碱下水解,采用绿色合成工艺:常温酶解得到去乙酰化产品,收率由78%提高到80%;最后进行胺甲基化反应,合成工艺也进行了工艺改进。文献[7]采用四氢呋喃作为反应溶剂,而此步的氯磺酸异氰酸酯对水及其敏感,四氢呋喃的工业回收具有较大的危险性,而且溶剂成本较高,改进后的工艺采用工业化溶剂丙酮,易于回收,成本较低,适合工业化生产。

1 实验部分
1.1 仪器和药品
Agilent1200/1260高效液相色谱仪,ARX-300型核磁共振仪,AB—HS型质谱仪(英国VG公司)。7-ACA(山东鲁抗医药股份有限公司),次氯酸叔丁酯自制,其他溶剂与试剂来源于上海国药集团。
1.2 合成过程
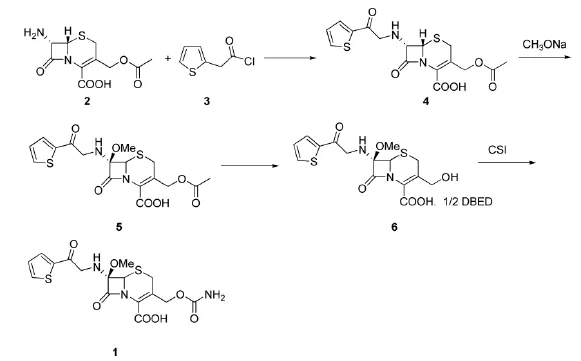
1.2.1化合物4(头孢噻吩酸)的合成
在1000 mL的三口烧瓶中加入7-ACA(68 g,0.25mol),三乙胺(50g,0.50 mol),二氯甲烷(300 mL,4.68 mol),水(15 mL,0.83 mol),30℃~35℃反应1 h,反应液澄清,将反应液冷却至-15℃~-10℃,缓慢滴加噻吩乙酰氯(41 g,0.28 mol),控制滴加时间2~3 h,滴加完毕,保温反应1 h,薄层监测反应完毕,往反应液中加入50 mL水,滴加36%盐酸调节在线pH计控制pH 2.0~2.5,搅拌20 min,静置分层;水层加200 g二氯甲烷萃取,合并有机层,加50 g饱和盐水洗涤,25 g无水硫酸镁干燥,过滤,滤液直接用于下步反应。
1.2.2化合物5(甲氧基头孢噻吩酸)的合成
往上述溶液中加入甲醇(50 mL,0.98 mol),将反应液保温-95℃~-90℃,滴加28%的甲醇钠的甲醇溶液(145 mL,1.31 mol)滴加完毕,保温滴加次氯酸叔丁酯(29 mL,0.28 mol)滴加完毕,保温反应30 min,反应完毕,往反应液中加入冰乙酸(15.5 mL,0.83 mol),然后将反应液缓慢加入250 g 10%的焦亚硫酸钠溶液,滴加36%盐酸在线pH计控制pH 2.0~2.2,搅拌分层,水层弃去,往有机层中滴加6%的碳酸氢钠溶液,pH计在线控制pH 7.0~7.2之间,搅拌20 min,静置分层,水层真空脱气除去二氯甲烷直接用于下步反应。
1.2.3化合物6(去乙酰化甲氧基头孢噻吩酸)的合成
往上述待用溶液中加入25 g去乙酰酯酶,保持温度20℃~25℃,滴加5%的碳酸氢钠溶液,在线pH计控制pH 7.0~7.5,反应至pH稳定不变,反应完毕,控温20℃~25℃,分批加入N,N'-二苄基乙二胺醋酸盐(40 g,0.11 mol)搅拌1h,降温至5℃保温1 h,抽滤,固体加入丙酮(250 mL,3.4 mol)室温打浆1 h,抽滤,40℃真空干燥得白色固体82 g,收率:65%(以7-ACA计)。
1.2.4头孢西丁的合成
往500 mL的三口烧瓶中加入化合物6(82 g,0.16 mol),丙酮(300 mL,4.08 mol),降温至-45℃~-50℃,滴加氯磺酸异氰酸酯(45 g,0.32 mol),保温反应1 h,反应完毕,加水20 mL,0℃~5℃反应1 h,抽滤,滴加15%碳酸钠溶液pH计在线控制调节pH 5.0~5.5,蒸出丙酮约200 mL,加水500 mL,加入活性炭10g,脱色0.5 h,抽滤,滤液保温20℃~22℃,滴加10%盐酸至浑浊,开始缓慢养晶至pH稳定,继续滴加盐酸,pH计在线控制pH 2.0~2.2,保温1 h,抽滤,用100 mL水洗,40℃干燥得白色固体57.5g,收率:84%。熔点149℃~150℃,质量分数90%。IR(KBr):3290、3188、3001、2960、1732 cm-1。吸光度A(262 nm):200。H-NMR(DMSOd6,400MHz)δ:3.054(m,2H),3.330(s,3H),3.797(d,2H),4.697(s,1H),4.826(s,2H),4.955(m,1H),6.963(m,1H),7.352(m,1H),9.341(s,1H)。MS:m/z:425.5[M+H]。与文献[6]相符。
2 结论
以便宜易得的7-ACA为起始原料,采用改进工艺合成头孢噻吩酸,收率由81.7%提高到95%,考虑到工业化生产的简单化,头孢噻吩酸及甲氧基化产物不需要结晶,直接进行下步反应,采用常温酶解得到去乙酰化产品,收率由78%提高到80%,最后进行胺甲基化得到目标产品,工艺总收率由45.8%提高到54.6%,纯度也由90%提高到99.2%,工艺路线操作简便,节约生产成本,适合工业化生产。
[1]徐学儒.头孢菌素临床应用进展[J].医学理论与实践,1995,8(6):241-243.
[2]张致平.内酰胺抗生素研究的进展[J].中国新药杂志,2002,11(1):61-67.
[3]付明耀.头孢菌素进展[J].中国冶金工业医学杂志,2003,20(3):172-174.
[4]Christensen B G,Cama L D,Karady S,et a1.7-a-Methoxy cephalosporina:US,4297488[P].1981-10-27.
[5]Deshpande P B,Khadangale B P.Process for the preparation of cefoxitin:US,2006252928[P].2006-11-09.
[6]谭瑞明,张黎辉,叶澄海.抗菌药物头孢西丁的制备工艺: CN,101007812A[P].2007-08-01.
[7]王艳峰,吴国颖,裴文.头孢西丁合成工艺的改进[J].化工生产与技术,2010,17(4):25-27.
[8]潘春越,杜鹏,彭东明.头孢西丁钠的合成[D].长沙:中南大学,2007.
Improved Synthesis of Cefoxitin
HUANG Wei-ping,YU Zheng-ye,HE Fa-ming,HE Xiao-peng,JIN Li-kuo
(Zhejiang Dongbang Pharmaceutical Co.,Ltd.,Linhai,Zhejiang 317016,China)
Starting from 7-ACA,the cefoxitin was obtained via four steps including acetylation,meoxylation,deacetylation and carbamoylation.The product of acetylation and methoxylation could be used for methoxylation and deacetylation without erystallition and purification,and the materials were reduced and production cycle was shorten.The method is simple and convenient and can be scaled up to industrial production.
process impro vement;cefoxitin;synthesis
1006-4184(2015)4-0019-04
2014-10-08
国家火炬计划项目(编号:2013GH020628)。
黄伟平(1978-),男,湖北鄂州人,博士,工程师,主要从事药物合成研究与开发。E-mail:huang0004@126.com。
猜你喜欢
世界农药(2022年10期)2022-11-10
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
当代化工研究(2022年11期)2022-06-27
中国化妆品(2022年3期)2022-03-26
能源化工(2021年2期)2021-12-30
合成化学(2020年3期)2020-04-10
科海故事博览·中旬刊(2020年3期)2020-03-15
农药科学与管理(2019年8期)2019-11-23
安徽化工(2018年5期)2018-10-23
中成药(2017年4期)2017-05-17
