
液相色谱-质谱联用法测定人血浆中法莫替丁胶囊含量及其生物等效性评价
2015-11-19 05:16:46何承霖庄晓珺梁建英
复旦学报(医学版) 2015年4期
何承霖 陆 烨 庄晓珺 梁建英
(1复旦大学药学院药物分析教研室 上海 201203;2复旦大学附属妇产科医院药剂科 上海 200011)
法莫替丁属于第3代组胺H2受体拮抗剂,具有拮抗胃黏膜壁细胞的组胺H2受体并抑制胃酸分泌的作用,与其结构类似物雷尼替丁或西米替丁相比,具有更好的疗效[1]。目前临床上主要用于治疗胃溃疡、十二指肠溃疡以及由胃酸分泌过多所引起的疾病。根据相关文献报道,现已有数种常见的血浆中法莫替丁含量的测定方法,包括毛细管电泳法[2-3],高效液相色谱-紫外检测法[4]以及液相色谱-质谱联用法[5-6]等。文献中血样前处理大多采用液液提取或固相萃取,方法相对繁琐;样品测定采用HPLC法,灵敏度较差;报道的液质联用法样品制备繁琐[7]或者基质效应较大[8]。本文以甲醇沉淀蛋白上清液用水1∶1稀释,液相色谱-质谱联用法测定血浆中法莫替丁的含量,方法更加准确、快速。运用建立的方法,以上海信谊万象药业股份有限公司生产的法莫替丁胶囊为参比制剂,对福建三爱药业有限公司研制的法莫替丁胶囊进行相对生物利用度及其生物等效性评价的研究。通过测定23名健康志愿者交叉口服给药后的经时过程的血药浓度,对计算所得的相对生物利用度以及有关药动学参数cmax、tmax、AUC0-t、AUC0-∞进行统计分析处理并作出是否具有生物等效性的评价。
材料和方法
药品与试剂 试验胶囊(T)(法莫替丁胶囊,规格:20 mg,批号:120601,福建三爱药业有限公司);参比胶囊(R)(法莫替丁胶囊,规格:20 mg,批号:51120105,上海信谊万象药业股份有限公司);法莫替丁对照品(批号:10305-0001,中国生物制品检定所);内标盐酸雷尼替丁(批号:100163-2041006,中国生物制品检定所);甲醇(HPLC级,批号:1586807117,德国默克公司);甲酸(批号:14105,北京迪马科技公司);去离子纯水均取自 Millipore Direct-Q纯净水发生器。
仪器 API-3000质谱检测器(美国Applied Biosystems公司)以及 Analyst色谱工作站(1.4.1版本,美国Applied Biosystems公司),Agilent公司1100型高效液相色谱仪,包括G1312A四元泵,G1316A柱温箱,G1379A在线脱气机和CTC HTS型控温自动进样器(瑞士CTC Analytics公司);离心机:型号TGL-16G型离心机,上海安亭科学仪器厂;旋涡混合器:型号KW-80A,上海医科大学仪器厂。
色谱条件 色谱柱为Venusil XBP苯基柱(100 mm×2.1 mm,5μm);流动相为0.1%甲酸水溶液-甲醇(体积分数为60∶40);流速为0.30 mL/min;柱温为40℃;进样量为10μL。
质谱条件 离子源为电喷雾电离源(Electronic Spray Ion,ESI);正离子方式检测;扫描方式为多反应监测(MRM);喷射电压为5 000 V;离子源温度为500℃;气帘气流速为10 L/min,雾化气流速为8 L/min,碰撞气流速为4L/min,扫描时间为0.2 s。
LC/MS/MS条件的优化 采用正离子方式检测,按拟订的优化程序分别优化法莫替丁与雷尼替丁的测定条件,在一级全扫描质谱图中得到法莫替丁的准分子离子峰(Q1)[M+H]+(m/z:338.3),然后选择性对其进行二级质谱分析得到主要碎片离子(Q3)(m/z:189.2),最后再通过法莫替丁的Q1/Q3对CE、DP等参数逐一优化。内标物雷尼替丁的LC/MS/MS条件优化步骤与法莫替丁相同。用于定量和定性分析的质谱参数见表1。

表1 质谱参数表Tab 1 Parameters of mass spectrum
血浆样品处理 精密移取血浆200μL,置于1.5 mL EP管内,加入20μL内标溶液与800μL甲醇涡旋1 min,于12 000×g离心10 min,取上清液1∶1纯水稀释后进样10μL,进行 LC/MS/MS分析。
药动学试验
受试者的选择 本试验选择24名男性健康志愿受试者,年龄为(25.42±3.72)岁,身高为(173.04±4.12)cm,体重为(67.08±4.22)kg,体重指数(body mass index,BMI)为18~24。经询问病史并进行临床检查及生化检查证实心、肺、肝、肾功能均正常,无消化道、代谢异常病史,无神经系统疾病史及过敏史,无药物依赖史,身体健康志愿参加法莫替丁生物利用度试验。受试者在试验前2周至整个试验期间禁服任何其他药物,并签署知情同意书。后1人因自身原因退出试验,因此实际完成试验人数为23人。

图1 空白血样色谱图Fig 1 Chromatograms of blank plasma
试验方案 24名男性健康志愿受试者禁食过夜(10 h以上),于次日早晨单剂量空腹服用等剂量试验胶囊或参比胶囊40 mg,用250 mL温开水送服。本试验采用随机、单盲、交叉试验的方法,将24名受试者随机编号分为两组,交叉口服给药进行试验。血样采集前,埋置肝素留置针头,服药前取空白血样(0h),给药后于0.5、1、1.5、2、2.5、3、3.5、4、6、8、12、16 h共13个时间点分别于前臂肘正中静脉采集静脉血3 mL,收集于加有肝素的离心管内,4 000×g离心10 min,分离出血浆置于-20℃冰箱中保存。用药期间和用药过后,受试者均未发生任何不良反应。
统计学处理 采用DAS 2.1.1软件对测得的数据(cmax、AUC0-t、AUC0-∞等)进行计算及统计学处理。
结 果
方法学验证
分析方法特异性 在本试验条件下,法莫替丁的保留时间约为1.30 min,内标(雷尼替丁)的保留时间约为1.38 min,血浆中内源性物质与测定药物分离完全,不干扰药物测定。其典型图谱见图1~3。
标准曲线与线性范围 取若干只1.5 mL EP管,各加入空白血浆190μL,加入法莫替丁对照品溶液10μL,使血浆中药物浓度分别为2.5、5.0、10.0、25.0、50.0、100.0、250.0ng/mL,每个浓度5管,按“血样处理”项下的方法操作后,作LC/MS/MS分析。以样品峰面积(Ai)与内标峰面积(As)比值对药物浓度(C,ng/mL)进行线性回归,计算结果为Y=0.002 102X+0.001 837(r=0.999 9,n=7)。线性范围:2.5~250 ng/mL。

图2 空白血样加法莫替丁与内标雷尼替丁的色谱图Fig 2 Chromatograms of control of Famotidine and control of Ranitidine
图3 志愿者血样加内标雷尼替丁色谱图Fig 3 Chromatograms of sample plasma with Ranitidine
精密度试验 配制定量限、低、中、高4个点(2.5、5.0、50.0、250.0ng/mL)的标准血样各6份,于同一天按血样预处理的方法操作后进样10μL,作LC/MS/MS分析,计算批内精密度连续3天,同法作LC/MS/MS分析,综合3天的数据,计算批间精密度,结果见表2。
准确度试验-方法回收率 取健康人的空白血浆190μL若干,分别加入法莫替丁对照品溶液,使法莫替丁浓度分别为2.5、5.0、50.0、250.0ng/mL,再按“血样预处理”项下的方法操作后,作LC/MS/MS分析。将药物峰面积与内标峰面积比值代入标准曲线的方程中,计算所得的浓度除以理论浓度,即得方法回收率,分别为99.64%、97.23%、99.18%、99.47%,计算RSD,结果见表2。
提取回收率试验 取健康人空白血浆190μL数份,分别加法莫替丁对照品溶液,使血浆中法莫替丁的浓度分别为5.0、50.0、250.0ng/mL,按血样预处理后作LC/MS/MS分析,记录药物峰面积。另取相同量的法莫替丁对照液,用流动相稀释后,进相同量的法莫替丁,记录峰面积,计算提取回收率,分别为89.29%、93.37%、87.10%,计算 RSD,结果见表2。
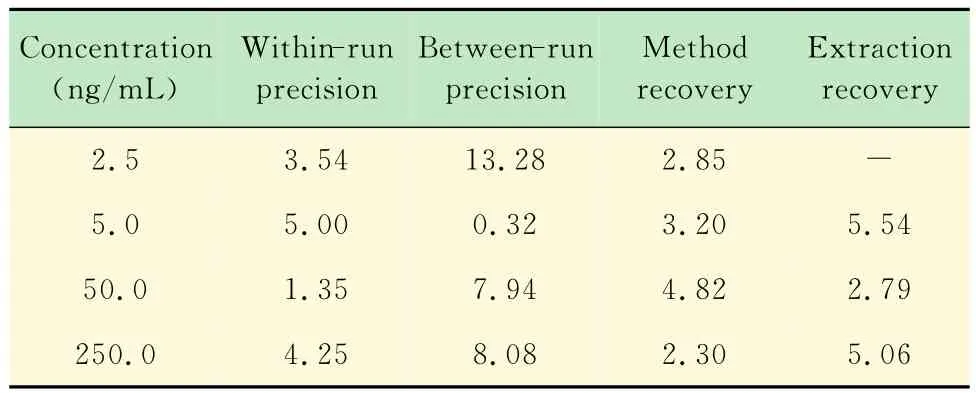
表2 方法学验证结果Tab 2 Result of method validation (RSD%)
结果表明该法莫替丁血样测定方法准确度高,精密度好,适用于临床研究中法莫替丁血样的分离分析。
样品稳定性考察
样品冰冻稳定性 取健康人空白血浆190μL数份,分别加法莫替丁对照品溶液,使血浆中法莫替丁的浓度分别为5.0、60.0、300.0ng/mL,放置在-20℃冰箱中,分别于冷冻后的0、30及45天后取出测定,计算RSD,结果见表3。
样品冻融稳定性 取健康人空白血浆190μL数份,分别加法莫替丁对照品溶液,使血浆中法莫替丁的浓度分别为5.0、60.0、300.0ng/mL,放置在-20℃冰箱中,然后分别冻融3次测定其峰面积比值,计算RSD,考察其冻融稳定性,结果见表3。
复溶溶液稳定性 取5.0、60.0、300.0ng/mL 3种浓度的复溶溶液数份,室温放置,并分别于0、8、24 h测定其峰面积比值,计算RSD,结果见表3。

表3 样品稳定性考察结果Tab 3 Stability of samples (RSD%)
结果表明法莫替丁血样冰冻稳定性及冻融稳定性良好,复溶溶液在24 h内可保持稳定。
基质效应考察 取空白血浆直接经甲醇蛋白沉淀前处理,取上清液,即得空白基质溶液。在50%甲醇溶液和经过处理的空白基质溶液中精确加入一定量的混合标准溶液,配制成低、中、高3种浓度测试溶液,取10μL进样,按上述色谱条件进行分析,比较甲醇溶液和空白基质溶液中被测组分的峰面积,计算基质效应。
各被测组分的基质效应结果见表4,可知各浓度血样基质效应为89.01%~95.73%,基质效应较小。

表4 各浓度样品的基质效应Tab 4 Matrix effect of samples (n=5)
测定方法的质量控制 取健康人空白血浆190 μL数份,分别加法莫替丁对照品溶液,使血浆中法莫替丁的浓度分别为5.0、50.0、250.0ng/mL,其余同血样的预处理项下步骤操作,作为质控样品。每天志愿者血样测定时,随行进行质控样品测定,以考察方法可靠性。
药动学参数及生物等效性评价
药-时曲线 23名受试者分别口服法莫替丁的参比胶囊和试验胶囊后,血浆中法莫替丁的平均血药浓度-时间曲线见图4。
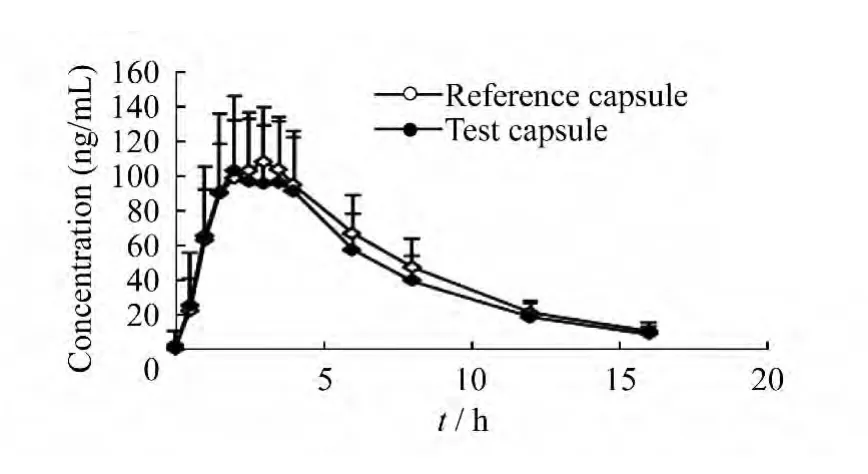
图4 23名健康受试者口服法莫替丁参比胶囊或试验胶囊的药-时曲线Fig 4 Plasma concentrations-time curve of Famotidine after oral administration of Famotidine reference capsules and tested capsules in 23healthy subjects
药动学参数 本实验采用DAS 2.1.1软件进行数据处理,23名受试者口服法莫替丁参比胶囊或试验胶囊后的药动学参数见表5。
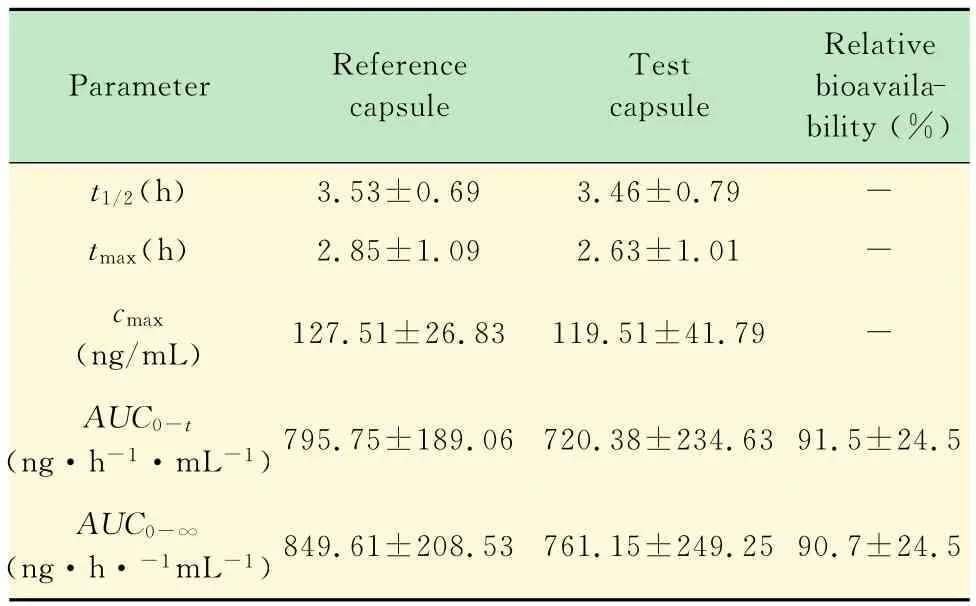
表5 23名健康受试者口服法莫替丁参比胶囊和试验胶囊后的药动学参数Tab 5 Pharmacokinetic parameters of Famotidine after oral administration of Famotidine reference capsules and tested capsules in 23healthy subjects
生物等效性分析 经方差分析,两种胶囊cmax、AUC0-t、AUC0-∞药剂间、周期间的F 值均小于相应的F临界值,说明两者在药剂间与周期间无显著性差异;双向单侧t检验和(1-2α)置信区间法计算所得结果表明,两种胶囊cmax、AUC0-t、AUC0-∞的tL≥t(1-0.05)与tH≥t(1-0.05)同时成立,并且试验胶囊和参比胶囊 AUC0-t几何均值比的90%CI(80.7%~97.4%)以 及 AUC0-∞的 90%CI(80.0% ~96.5%)均落在等效标准范围内(80%~125%),cmax几何均值比的90%CI(80.4%~100.6%)也落在等效标准范围内(75%~133%),试验胶囊与参比胶囊的tmax经非参数法检验,差异无统计学意义(P>0.05)。因此,生物等效性分析结果表明两种胶囊间主要药物代谢动力学参数差异无统计学意义,即两者具有生物等效性。
讨 论
法莫替丁分子结构中含有一强极性胍基,水溶性较大,文献中即使采用液相色谱质谱联用测定样品,前处理亦大多采用有机溶剂提取或固相萃取法[7],前处理所需时间较长且操作繁琐。本文采用甲醇以水相与有机相1∶4沉淀蛋白,方法简便快速,可在约10 min时同时处理多达12份血样。同时在进样前用纯水1∶1稀释上清液,与同样采用蛋白沉淀液质联用的测定方法[8]相比大大降低了试验的基质效应,低、中、高3个浓度的基质效应可达到89.01%~95.73%,而且也能得到令人满意的提取回收率,平均可达85%以上,提取效果优于已有的文献报道[8]。本试验操作简单快速,样品的分析时间较短,可满足临床研究中大量样品的快速分析。
另外,本文选择结构与法莫替丁比较相似的雷尼替丁作为内标,在样品的前处理过程中具有相近的提取回收率,可以为定量方法提供更好的准确度和精密度。
[1] Howden CW,Tytgat GN.The Tolerability and safety profile of famotidine[J].Clinical Therapeutics,1996,18(1):36-54.
[2] Helali N,Tran NT, Monser L.Capillary zone electrophoresis method for the determination of famotidine and related impurities in pharmaceuticals[J].Talanta,2008,74(4):694-698.
[3] Pérez-Ruiz T,Martinez-Lozano C,Tomás V.Direct determination of ranitidine and famotidine by CE in serum,urine and pharmaceutical formulation[J].J Pharm Biomed Anal,2002,30(4):1055-1061.
[4] Arayne MS,Sultana N,Zuberi MH.Simultaneous determination of metformin,cimetidine,famotidine,and ranitidine in human serum and dosage formulations using HPLC with UV detection[J].J Chromatogr Sci,2010,48(9):721-725.
[5] Wang X,Rytting E,Abdelrahman DR.Quantitative determination of famotidine in human maternal plasma,umbilical cord plasma and urine using high-performance liquid chromatography-mass spectrometry[J].Biomed Chromatogr,2013,27(7):866-873.
[6] Zhong L,Eisenhandler R,Yeh KC.Determination of famotidine in low-volume human plasma by normal-phase liquid chromatography/tandem mass spectrometry[J].J Mass Spectrom,2001,36(7):736-741.
[7] 桂静,杜青青,王凌.人血浆中法莫替丁的测定及其药物动力学的研究[J].华西药学杂志,2010,25(2):147-150.
[8] 翁静艳,杭太俊,张宏文.复方法莫替丁咀嚼片的人体药动学[J].中国新药与临床杂志,2006,25(7):523-526.
猜你喜欢
中国心血管杂志(2022年2期)2022-11-25 17:29:20
中国心血管杂志(2022年4期)2022-11-25 16:59:06
中国典型病例大全(2022年9期)2022-04-19 21:26:28
东坡赤壁诗词(2021年1期)2021-03-24 18:25:36
中国心血管杂志(2021年6期)2021-01-02 08:18:16
中国心血管杂志(2019年3期)2019-01-04 16:25:09
中国医药指南(2017年3期)2017-11-13 02:56:05
护士进修杂志(2017年1期)2017-02-28 19:49:40
甘肃畜牧兽医(2016年20期)2016-03-12 03:59:00
四川畜牧兽医(2014年9期)2014-12-31 12:30:58
