
H2和CO2在金属-有机架构(MOFs)材料中的吸附性质
2015-11-17 12:11孙迎新毛新峰
应用技术学报 2015年1期
孙迎新, 毛新峰
(上海应用技术学院化学与环境工程学院,上海 201418)
H2和CO2在金属-有机架构(MOFs)材料中的吸附性质
孙迎新, 毛新峰
(上海应用技术学院化学与环境工程学院,上海 201418)
使用巨正则系综蒙特·卡洛(GCMC)方法以及TEAM力场参数考察了H2和CO2分子在IRMOF-1和IRMOF-16两种晶体材料中的吸附性质,用统计涨落理论计算了客体分子的吸附热,并分析了柔性骨架和刚性骨架对客体分子吸附性质的影响.计算表明,柔性和刚性骨架对吸附量和吸附热影响可以忽略.客体分子的体积吸附量在IRMOF-1中更大,重量吸附量在IRMOF-16中更大.IRMOF-16的吸附热小于IRMOF-1.这些结论都与前人的实验及理论研究相吻合.
TEAM力场;吸附量;柔性骨架;吸附热
近年发展起来的纳米多孔金属-有机架构(metal-organic frameworks,MOFs)材料引起了人们广泛的研究兴趣.MOFs的应用领域主要涉及气体储存、化学分离、催化过程等[1].这种材料属于配位聚合物,是由无机金属团簇或无机氧化物团簇与各种类型的有机官能团(主要是聚合羧酸盐)空间自组装而成,形成各种不同周期性结构的晶体.2002年,美国化学家Yaghi在Science杂志公布,实验上已经合成出了一系列具有相同网状结构的IRMOFs(isoreticular MOFs)晶体,如IRMOF-1~IRMOF-8,IRMOF-10,IRMOF-12,IRMOF-14,IRMOF-16,这些MOFs材料表现出了很好的甲醇储存吸附性能,有良好的工业应用前景[2].近年来,H2、N2、O2、CO2和C6H6等客体分子在MOFs中的吸附和扩散性质越来越受到人们的重视,这些研究有望缓解能源紧缺和环境污染治理问题.
从实验角度而言,合成具有特定功能和性质的MOF材料不是一件容易的事,而分子模拟方法由于能够预测诸如分子的吸附、扩散等性质,使得人们可以在实验室合成之前就能了解其潜在的合成价值.这些模拟方法中,分子动力学、蒙特·卡洛方法能够直接预测材料吸附量和客体分子的扩散系数,非常适合处理大分子体系.针对MOF材料吸附H2和CO2气体,已有很多相关工作[3-6].如对实验上已经合成的IRMOF-1和IRMOF-16晶体,就有很多相关理论计算成果.Yang等[3]研究了包括IRMOF-16在内的5种IRMOFs对CO2/N2混合气体的分离效果,发现一种被称为阶梯型吸附的分离现象,并认为这种阶梯型吸附能够提高混合物中CO2的分离效率.Garberoglio等[4]使用蒙特·卡洛方法计算了H2、He、甲烷等气体在各种典型MOFs,如IRMOF-1、IRMOF-6、IRMOF-14、Cu-BTC中的吸附量,发现H2在室温下吸附量很低,没有一种MOF能用来充当美国能源部电动汽车用的氢气储存系统.Getman等[5]考察了金属离子改性后的IRMOF-1、IRMOF-16等5种MOFs材料的储氢性能,发现Mg改性的材料能大大提高室温下MOFs的H2吸附量.Fu等[6]用蒙特·卡洛方法研究了IRMOF-1、IRMOF-16等9种MOFs在77、200、298 K条件下对氢气的吸附量和吸附热,所得计算结果与实验值较为吻合.Skoulidas等[7]研究了H2、CO2、CH4、N2等在IRMOF-1,Cu-BTC等材料中的吸附等温线和扩散系数,得到了一些与实验相符合的结论.这些代表性工作中,大多用的是刚性骨架和标准力场,不是自己开发的力场,适用性受到限制,而分子模拟的核心是分子力场.本课题组在前期已经开发出一套针对IRMOFs的TEAM力场,这套力场可以准确预测各种IRMOFs晶体的机械性质、弹性性质、振动光谱和客体分子在其中的扩散性质[8].本文拟解决的关键问题是:用这套力场考察H2和CO2分子在两种典型的晶体,IRMOF-1和IRMOF-16中的吸附性质,解决诸如吸附量和吸附热、柔性和刚性骨架对吸附性质的影响等关键科学问题,这些问题是考察一个力场正确性以及应用范围的重要标准.一些较大尺寸的分子在材料中吸附时会对材料骨架产生膨胀、收缩以及扭曲等影响,这些影响必须通过柔性骨架模型才能描述出来,而柔性骨架的模型只能用包含键参数的柔性力场来描述,而不能用只包含非键参数的刚性力场来描述.用柔性力场和柔性骨架来描述H2和CO2分子在IRMOF-1和IRMOF-16中的吸附性质,可清楚地再现客体分子对材料骨架的影响因素,并由此得到规律性的结论.
1 模拟方法
模拟中采用广泛使用的可迁移性(transferable)、可扩展性(extensible)、准确性(accurate)和模块性(modular)的TEAM力场[9]函数形式:

式中:

所有的MOFs骨架原子参数来源于文献[8]中的力场参数.非键范德华(VDW)相互作用采用伦纳德-琼斯(Lennard-Jones(12-6),LJ-12-6)形式,不同原子对之间的参数采用Lorentz-Berthelot混合规则,即:

对于客体分子H2和CO2,分别采用Darkrimand Levesque(DL)[10]和Murthy-Singer-McDonald[11]三点刚性模型.对H2分子,在分子中间位置有1个虚原子,H—H键长固定为0.074 1 nm,H原子和虚原子所带电荷分别为+0.466 4e和-0.932 8e,H原子无LJ参数,虚原子的LJ参数为:σ=0.295 8 nm,ε=36.68 K.对CO2分子,固定C—O键键长为0.118 nm,O—C—O键角为180°,C和O原子电荷分别为+0.576e和-0.288e,LJ参数为:σC=0.278 9 nm,εC=29.66 K,σO=0.301 1 nm,εO=82.96 K.
模型采用IRMOF-1和IRMOF-16两种晶体,晶体结构如图1所示.它们分别属于Fm-3m和Pm-3m空间群,晶体的原子坐标参考文献值[2],计算中使用2×2×2晶格,用周期性边界条件消除有限边界效应,静电相互作用使用Ewald加和方法计算,VDW相互作用能的半径截断值为1.6 nm,并使用尾部校正方法(tail corrections)消除能量计算误差.模拟方法使用巨正则系综蒙特·卡罗(GCMC)方法,考察了在77、233、298 K,以及0~10 MPa范围内晶体对客体分子的吸附量以及吸附热.同时考察柔性骨架和刚性骨架对吸附量和吸附热的影响,分别遵循如下公式:
重量吸附量

体积吸附量
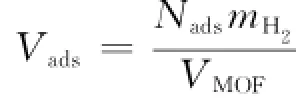
吸附热使用统计力学涨落理论来计算
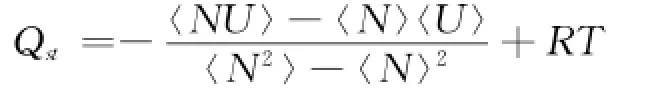
式中:尖括号表示系综平均;N和U表示吸附的客体分子数和系统的总能量;T表示系统温度.
对每一次GCMC模拟,平衡步骤包括了2× 106次原子或分子移动,并用相同的移动次数进行统计取样.原子或分子的移动(即moves)包括客体分子在MOF晶体中的插入、删除、平动和转动,在柔性骨架中还包括了MOF晶体本身原子的平动.客体分子的化学势采用Widom插入法计算,整个模拟过程采用Towhee 7.0.6软件进行[12].
图1 晶体结构图Fig.1 Structure charts of crystals
2 结果与讨论
2.1 客体分子在柔性MOFs骨架中的吸附量对比
在77、233、298 K下H2分子在IRMOF-1和IRMOF-16柔性骨架中的吸附量如图2所示.由图可见,对每一种MOF晶体,温度升高时,无论重量吸附量还是体积吸附量均降低.这主要是因为随着温度的增大,客体分子的热运动越来越剧烈,与晶体骨架的结合能力越来越弱,结合能越来越小,吸附热的数据将证实这一点.另一方面,由IRMOF-1与IRMOF-16对比可知,体积吸附量前者更大,重量吸附量后者更大,此现象与前人的结论一致[6].由于IRMOF-1晶体体积小于IRMOF-16体积,以晶体单胞作为基本单元,前者自由体积为13.862 52 nm3,后者为72.580 05 nm3,而且分子量方面前者大于后者,故IRMOF-16晶体密度更小,重量吸附量更大.但正是由于IRMOF-16的体积远大于IRMOF-1,从而使得单位体积内吸附的H2数目减少,故体积吸附量小于IRMOF-1.
8 MPa时,77 K和298 K两个温度下H2在两种MOFs中的重量吸附量数据见表1,计算值和文献实验值[13]以及其他理论值[6]都较为接近,说明计算结果的准确性较好.
298 K时,CO2分子在IRMOF-1和IR-MOF-16柔性骨架中的吸附量如图3所示.CO2分子和H2分子在两种晶体中表现出了相似的吸附性质,如,体积吸附量IRMOF-1更大,重量吸附量IR-MOF-16更大.在298 K及同样压力时,CO2的吸附量明显大于H2分子,这主要是因为CO2分子和晶体骨架的结合能更大,更容易吸附在晶体表面上.

图2 不同温度下柔性IRMOF-1和IRMOF-16骨架中H2分子的吸附量对比Fig.2 The comparison of adsorption capacity of H2in flexible IRMOF-1 and IRMOF-16 crystals at different temperatures
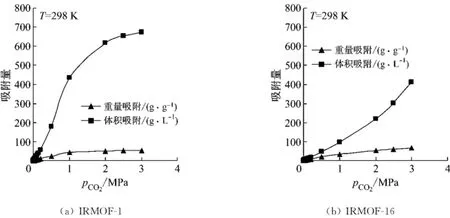
图3 柔性IRMOF-1和IRMOF-16骨架中CO2分子的吸附量对比Fig.3 The comparison of adsorption capacity of CO2in flexible IRMOF-1 and IRMOF-16 crystals
2.2 客体分子在柔性MOFs骨架中的吸附热对比
由于不同负载下客体分子的吸附热互不相同,选择了接近零负载情况下的吸附热进行对比,数据见表2.由表2可见,对于柔性MOF骨架,随着温度升高,H2分子吸附热下降,这是因为随着温度增大,客体分子热运动更剧烈,与晶体骨架的结合能力趋于减弱,结合能越来越小,反过来也会减少吸附量,上述吸附量的数据已经证明了这一点.对于H2和CO2分子,IRMOF-16的吸附热小于IRMOF-1,原因在于IRMOF-16的自由体积占总体积的比值(91.41%)大于IRMOF-1(80.42%),自由体积是晶体三维盒子中除去MOF骨架原子后的部分(包含了MOF骨架原子的范德华半径),自由体积比重越大,相当于空洞越大,被骨架吸附的客体分子比重就减少了,从而描述骨架和客体分子相互作用的吸附热就趋于减少,这与前人的计算结果一致[14].另外,CO2的吸附热明显大于H2,这是由于CO2的极化率大于H2的极化率.实验值方面,对于H2分子,77 K下的计算值高于实验值(IRMOF-1上约为5.0 kJ·mol-1)[15],这可能是低温下量子效应引起的.对于CO2,实验上用微量热法测得CO2分子在一种MIL-53(Al)型MOF晶体中300 K,0~10 MPa范围内的吸附热在10~40 kJ·mol-1[16],表2的计算值与此相符.

表1 不同温度下H2分子在IRMOF-1和IRMOF-16中的重量吸附量Tab.1 The gravimetric adsorption capacity of H2in IRMOF-1 and IRMOF-16 at different temperatures g/g
2.3 柔性骨架和刚性骨架对吸附性质的影响
刚性MOF骨架的吸附性质与柔性骨架的差别如图4所示,分别是77 K下H2和298 K下CO2在IRMOF-1中的吸附量,在IRMOF-16中趋势相同.无论重量吸附还是体积吸附,无论在IRMOF-1中还是在IRMOF-16中,柔性骨架和刚性骨架对H2和CO2的影响可以忽略不计.吸附热方面,由表2可看出,刚性骨架基本可以正确反映柔性骨架对吸附热的温度效应,差别也可以忽略.这主要是由于两种客体分子的尺寸较小,如果是体积较大的分子,如苯分子,柔性骨架将表现出不同,特别是扩散性质,这在已发表文献中有所证实[17].

表2 接近零负载时H2和CO2分子在IRMOF-1和IRMOF-16晶体中的吸附热Tab.2 The isosteric heat of adsorption at near zero loading of H2and CO2in IRMOF-1 and IRMOF-16 crystals kJ/mol

图4 柔性和刚性IRMOF-1骨架对H2和CO2分子吸附量的影响Fig.4 The effect of flexible and rigid IRMOF-1 frameworks on adsorption capacity of H2and CO2
3 结 语
本文考察了H2和CO2分子在柔性和刚性IRMOF-1和IRMOF-16两种MOF材料中的吸附性质.计算结果表明,柔性和刚性骨架对吸附量和吸附热影响可以忽略.对每一种MOF材料,无论重量吸附量还是体积吸附量,当温度升高时均降低.数据对比可知,客体分子的体积吸附量在IRMOF-1中更大,重量吸附量在IRMOF-16中更大.
吸附热方面,随着温度升高,客体分子热运动加剧,与晶体骨架的结合能力减弱,吸附热降低.同时,由于自由体积在晶体总体积中所占比重不同,导致自由体积比重更高的IRMOF-16吸附热小于IRMOF-1.这些结论都与前人的研究相吻合,说明了分子力学力场参数的精确性和可靠性较高,能够应用于气体分子的吸附性质研究.
[1] Kitagawa S,Noro S,Nakamura T.Pore surface en-gineering of microporous coordination polymers[J]. Chemical Communications,2006,7:701-707.
[2] Eddaoudi M,Kim J,Rosi N,et al.Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage[J]. Science,2002,295(18):469-472.
[3] Yang Q Y,Ma L L,Zhong C L,et al.Enhancement of CO2/N2mixture separation using the thermodynamic stepped behavior of adsorption in metal-organic frameworks[J].The Journal of Physical Chemistry C,2011,115(6):2790-2797.
[4] Garberoglio G,Skoulidas A I,Johnson J K.Adsorption of gases in metal organic materials:Comparison of simulations and experiments[J].The Journal of Physical Chemistry B,2005,109(27):13094-13103.
[5] Getman R B,Miller J H,Wang K,et al.Metal alkoxide functionalization in metal-organic frameworks for enhanced ambient-temperature hydrogen storage[J].The Journal of Physical Chemistry C,2011,115(5):2066-2075.
[6] Fu J,Sun H.An ab initio force field for predicting hydrogen storage in IRMOF materials[J].The Journal of Physical Chemistry C,2009,113(52):21815-21824.
[7] Skoulidas A I,Sholl D S.Self-diffusion and transport diffusion of light gases in metal-organic framework materials assessed using molecular dynamics simulations[J].The Journal of Physical Chemistry B,2005,109(33):15760-15768.
[8] Sun Yingxin,Sun Huai.An all-atom force field developed for Zn4O(RCO2)6metal organic frameworks[J].Journal of Molecular Modeling,2014,20(3):2146-2160.
[9] Sun H.Prediction of fluid densities using automatically derived VDW parameters[J].Fluid Phase Equilibria,2004,217(1):59-76.
[10] Darkrim F,Aoufi A,Malbrunot P,et al.Hydrogen adsorption in the Na A zeolite:A comparison between numerical simulations and experiments[J].The Journal of Chemical Physics,2000,112(13):5991-5999.
[11] Murthy C S,Singer K,McDonald I R.Interaction site models for carbon dioxide[J].Molecular Physics,1981,44(1):135-143.
[12] Martin M G.MCCCS Towhee[EB/OL].(2006-07-11)[2014-05-22].http://towhee.sourceforge.net/.
[13] Kaye S S,Dailly A,Yaghi O M,et al.Impact of preparation and handling on the hydrogen storage properties of Zn4O(1,4-benzenedicarboxylate)3(MOF-5)[J].Journal of the American Chemical Society,2007,129(46):14176-14177.
[14] Cao D P,Lan J H,Wang W C,et al.Lithium-doped 3D covalent organic frameworks:High-capacity hydrogen storage materials[J].Angewandte Chemie International Edition,2009,48(26):4730-4733.
[15] Kaye S S,Long J R.Hydrogen storage in the dehydrated prussian blue analogues M3[Co(CN)6]2(M =Mn,Fe,Co,Ni,Cu,Zn)[J].Journal of the American Chemical Society,2005,127(18):6506-6507.
[16] Ramsahye N A,Maurin G,Bourrelly S,et al. Charge distribution in metal organic framework materials:Transferability to a preliminary molecular simulation study of the CO2adsorption in the MIL-53(Al)system[J].Physical Chemistry Chemical Physics,2007,9:1059-1063.
[17] Greathouse J A,Allendorf M D.Force field validation for molecular dynamics simulations of IRMOF-1 and other isoreticular zinc carboxylate coordination polymeters[J].The Journal of Physical Chemistry C,2008,112(15):5795-5802.
(编辑 吕丹)
Adsorption Properties of H2and CO2in Metal-Organic Frameworks(MOFs)Materials
SUN Ying-xin, MAO Xin-feng
(School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai 201418,China)
The adsorption properties of H2and CO2on IRMOF-1 and IRMOF-16 crystals using grand canonical Monte Carlo(GCMC)method and TEAM force field parameters were investigated.The isosteric heat of adsorption for guest molecule was calculated by statistical fluctuation theory.The effect of flexible and rigid frameworks on adsorption properties was studied.Simulation results suggested that effect of flexible and rigid frameworks on adsorption capacity and heat was negligible.The volumetric adsorption capacity of guest molecule in IRMOF-1 turned out to be higher than that in IRMOF-16,but the gravimetric adsorption capacity in IRMOF-1 was lower.The calculated isosteric heat for IRMOF-16 was lower than that for IRMOF-1.All results were in line with the previous experimental and theoretical studies.
TEAM force field;adsorption capacity;flexible framework;heat of adsorption
O 642.1
A
1671-7333(2015)01-0023-06
10.3969/j.issn.1671-7333.2015.01.004
2014-06-04
国家自然科学基金青年基金资助项目(21203118);上海市教委科研创新基金资助项目(14YZ147);上海市青年教师培育基金资助项目(ZZyyy12005);上海应用技术学院引进人才基金资助项目(YJ2012-11)
孙迎新(1980-),男,讲师,博士,主要研究方向为催化反应机理的QM/MM模拟及晶体材料的分子力学力场开发.
E-mail:sunyingxin0312@sit.edu.cn
猜你喜欢
中国音乐学(2022年1期)2022-05-05
当代陕西(2022年5期)2022-04-19
河南科学(2020年3期)2020-06-02
时代人物(2019年10期)2019-09-18
时代人物(2019年25期)2019-09-13
中财法律评论(2018年0期)2018-12-06
教学与管理(理论版)(2017年7期)2017-08-11
制造技术与机床(2017年3期)2017-06-23
专利代理(2016年1期)2016-05-17
浙江人大(2014年1期)2014-03-20
