
脊柱外周原始神经外胚层瘤CT、MRI及病理表现
2015-11-09 02:46牛富业马燕青邹薇薇徐才国尹雪军王晨光
中国医学计算机成像杂志 2015年2期
牛富业 马燕青 邹薇薇 徐才国 尹雪军 王晨光
原始神经外胚层肿瘤( primitive neuroectodermal tumor,PNET) 是一种罕见的起源于原始神经管胚基细胞、向原始神经分化的小圆细胞恶性肿瘤,按发病部位的不同,分为中央型( central primitive neuroectodermal tumor,cPNET)和外周型( peripheral primitive neuroectodermal tumor,pPNET)[1]。pPNET多发生于胸壁、四肢,脊柱较罕见[2]。本文回顾性分析13例经手术病理证实的脊柱pPNET患者临床、CT、MRI及病理学资料,旨在提高对本病诊断的准确性,为临床诊断、治疗提供依据。
方 法
1. 研究对象
收集2011年10月—2014年3月经我院手术病理证实,临床、影像学及病理资料完整的13例脊柱pPNET。其中女性5例,男性8例,年龄8~49岁,平均26.4岁。
2. 检查方法
患者均行CT和MRI检查。CT检查设备为Philips Brilliance 256层iCT,管电压120kV,管电流200mA,旋转时间0.5s,准直为0.625mm,层厚及层间隔均为5mm,采用1mm层厚进行重建。11例行增强检查,对比剂为碘海醇注射液(350mgI/ml,1.0ml/kg)经肘静脉以3.0ml/s注射45s后扫描。MR检查设备为Philips achieva 3.0T TX超导磁共振扫描仪,脊柱表面线圈,矩阵256×256,NEX 2,层厚3~4mm,层间距0~1mm。常规扫描:矢状位SE T1WI(TR508ms,TE12ms)、FSET2WI(TR4000ms,TE108ms),FOV 300mm;轴位FSE T2WI(TR4200ms,TE 108ms),FOV220~300mm。增强检查对比剂为钆喷替酸葡甲胺注射液(Gd-DTPA)0.1mmol/kg体重,经肘静脉以2.0ml/s注射20s后行抑脂轴位(TR550ms,TE9ms)、矢状位(TR 500ms,TE9ms)及冠状位(TR500ms,TE9ms)扫描。
3.影像学评价
由两位经验丰富的骨肌系统副主任医师以上影像学专家对图像进行独立阅片。分析发生病变椎体的水平,病灶大小、形态、边界,椎体及椎间孔有无受累,椎旁软组织及病灶的密度、信号及强化特点,有无囊变、坏死及钙化,病灶与周围脊髓及神经的关系。意见不同时经协商最后达成一致结果。
4.病理学检查
患者均手术切除病变,标本行常规石蜡包埋切片、HE及免疫组化染色。免疫组化主要检测 CD99、CD34、神经元特异性烯醇酶(NSE)、波形蛋白(Vim)、S100、CK和扁豆凝集素(LCA)。
结 果
1. 病变部位及临床表现
单发病灶10例(颈椎3例,腰椎2例,腰骶段1例,骶尾椎4例),多发病灶3例。10例位于椎体和/或附件,2例位于椎间孔,1例位于髓外硬膜下(图1)。8例病灶累及椎管内外,5例突入相应椎间孔,2例病灶累及骶髂关节及双侧髂骨。患者的临床表现主要为发病部位疼痛及病灶占位效应引起的神经压迫症状。2例伴有大小便障碍,4例伴有双下肢疼痛、乏力及麻木,2例伴有右上肢疼痛。2例双下肢肌张力减弱伴肌肉萎缩、精细动脉障碍,2例四肢肌张力减弱,2例四肢肌张力增强。5例伴有会阴区及双下肢触觉、痛温觉减低,1例右上肢触觉及痛温觉减退,2例双上肢、下肢及锁骨平面以下痛温觉减退。
2. CT、MR表现
CT及MR表现为骨质破坏伴浸润性生长的不规则软组织肿块,边界不清。肿块较大,平均最大径6.5cm,除1例位于髓外硬膜下的病灶较小,长径约2.1cm外(图1),其余长径均大于4cm。11例伴有椎体、附件骨质破坏,4例呈溶骨性骨质破坏,7例呈混合性骨质破坏,其中2例以成骨为主(图2A),5例以溶骨为主,2例未见明显骨质破坏。CT平扫为等或低密度(图3B),3例病灶内可见少量条片状钙化影。MR平扫与周围肌肉比T1WI为等或低信号(图1A、2A),T2WI为高或混杂高信号(图1B、2B)。增强扫描实性成分明显不均匀强化,坏死、囊变区未见明显强化(图1C、2C、3D)。8例患者累及椎管内外,脊髓受压,1例脊髓受侵(图2C);5例患者可见不典型哑铃征,通过扩大的椎间孔与椎外部分相连(图3D)。4例伴有转移,其中肺转移2例(图2D),骨转移3例。术前诊断恶性肿瘤7例,神经源性肿瘤5例,炎症1例。
3. 病理结果
肿块大体切面为灰白色或暗红色,呈鱼肉样或胶冻状,质韧,肿物与周围组织分界不清,可见不完整包膜,血管较丰富。镜下肿瘤细胞由呈巢状分布的小圆细胞构成,胞质疏松,核深染,可见不典型有丝分裂、核碎裂及Homer-Wright玫瑰结(图3E)。免疫组化显示有两项以上神经源性抗体呈阳性表达,肿瘤均表达CD99(图3F),Vim和S100多数阳性,LCA均为阴性(表1)。
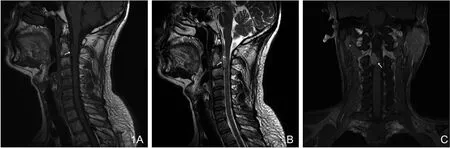
图1 男,49岁,椎管内PNET。颈2椎体水平髓外硬膜下长径约2.1cm的梭形软组织影(↑)。T1WI(A)呈等、低信号,T2WI(B)上呈混杂性稍高信号,增强扫描(C)可见不均匀明显强化,脊髓受压,椎间孔未见明显扩张。

图2 女,15岁,颈、胸椎多发PNET。颈2及胸1椎体骨质破坏,周围可见较大软组织肿块影并突入椎管内(↑),硬膜囊、脊髓明显受压;T1WI(A)呈等信号,T2WI(B)呈混杂高信号,增强扫描(C)可见明显不均匀强化,脊髓受侵,内可见结节状强化(↑)。胸部CT(D)两肺可见多发转移。
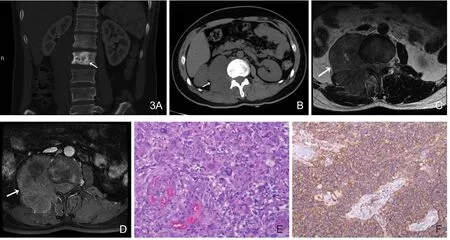
图3 男,24岁,腰椎PNET。腰1、2椎体呈以成骨(↑)为主的混合性骨质破坏(A),右侧椎旁可见巨大软组织密度肿块影(B),肿物向椎管内生长,包绕椎管,硬膜囊脊髓受压,可见不典型哑铃征(↑),T2WI呈混杂稍高信号,内可见囊变、坏死(C),增强扫描(D)可见明显不均匀强化。病理镜下肿瘤细胞由呈巢状分布的小圆细胞构成,胞质疏松,核深染,可见Homer-Wright玫瑰结(E,HE×100)。CD99呈阳性表达,细胞质和细胞膜呈弥漫性染色(F,En Vision×100)。

表1 13例脊柱外周原始神经外胚层瘤临床特点及免疫组化结果
讨 论
1. 概述
WHO将PNET定义为低或未分化细胞组成的胚胎性肿瘤,可向神经元细胞、室管膜细胞、星形细胞、肌细胞及黑色素谱系细胞分化[3]。pPNET、骨尤因肉瘤(Ewing’s sarcoma,Es)、骨外尤因肉瘤、Askin瘤因预后、生物学行为和组织上CD99的表达相似而合称为尤因家族肿瘤[4];Es在尤因肿瘤家族中分化最低,而pPNET分化最高[2]。
pPNET发病年龄多在30岁以下[5],儿童和青少年多见。全身各处均可发生,脊柱的pPNET罕见,在原发性脊柱肿瘤中所占比例不足l%,目前国内外文献多为个案报道[6-7]。本组13例中有9例发病年龄在30岁以下,最小8 岁,与文献报道相符。
2. CT和MRI表现
脊柱pPNET无明显特征性影像学表现,多表现为边界不清的软组织肿块及骨质破坏,软组织肿块及骨质破坏均较大[8];肿块边缘可见分叶或融合,周围组织明显受压,边界不清,表现其恶性特征[9]。CT平扫肿瘤多表现为以软组织密度为主的混杂密度;MR上肿瘤在T1WI较周围肌肉比为低或等、低信号,由于肿瘤生长较快,中央部分血供不足而易引起缺血性坏死、囊变,T2WI多为不均匀高信号;增强扫描肿瘤实性成分明显不均匀强化,囊变、坏死区无强化。文献[10]报道肿块中多无钙化和骨化,而本组病例中有7例可见成骨,其中有2例以成骨为主,3例软组织内可见钙化,发生机制尚需进一步研究。累及椎管内外的pPNET较易累及邻近椎间孔,致椎间孔扩大及邻近骨质破坏,形成不典型哑铃征。
3. 病理学特征
光镜下PNET由均匀一致的小圆细胞组成,细胞排列紧密,纤维蛋白的基质周围环绕形成Homer-Wright玫瑰花结,核内有丝分裂活跃,胞质成分少,核浓染,核质比例高。电镜下,细胞非常原始,细胞器稀少,胞质内有大量糖原,可见交织的神经突起和神经内分泌颗粒(100~150㎜)的超微结构[11]。组织学上,PNET与Es不能区别,鉴别诊断主要依靠免疫组化及电镜显示的不同神经分化特征。免疫组化显示有两项以上神经源性抗体呈阳性表达,神经源性抗体CD99有一定特异性,它是MIC2基因编码的一种p30-32跨膜糖蛋白,可出现于骨外Es和pPNET,但在cPNET中缺乏[2]。第三版(2002)WHO骨肿瘤分类中将尤因肉瘤和PNET从骨髓瘤中独立分类,称Es/PNET,此类下只有一个病种,即尤因肉瘤,而PNET包括其中[12]。两者免疫组化均表达CD99和NSE,细胞遗传学研究证实两者均存在频发性、非随机性染色体易位t(11;22)(q24;q12)。第四版(2013)WHO骨肿瘤分类中未延续旧版中将Es与PNET并列,而是将PNET删除,但在Es组织病理学陈述中分为经典型、非典型型及PNET型,提示Es与PNET形态上的相似性[13]。
4. 鉴别诊断
脊柱pPNET需与脊柱转移瘤、淋巴瘤、嗜酸性肉芽肿、神经源性肿瘤等鉴别。转移瘤与pPNET在影像学上表现相似,但转移瘤发病年龄较大,有原发肿瘤病史,病灶常多发,跳跃式骨质破坏。淋巴瘤患者年龄较大,可有全身浅表淋巴结肿大、肝脾肿大等。淋巴瘤信号多较均匀,坏死囊变少见,常呈浸润性生长,侵犯椎管有包绕脊髓呈纵形生长的趋势。脊柱嗜酸性肉芽肿青少年多见,多发生于胸椎和腰椎。患椎主要表现为囊状或溶骨性骨质破坏,轻度膨胀,边缘多不规则,可有硬化,破坏区内可见骨嵴。晚期椎体常发生明显病理性压缩,呈楔状或高度致密的平板状改变,其横径及前后径均超出正常椎体[14]。累及椎管内外的pPNET需与神经源性肿瘤鉴别,后者为最常见的髓外硬膜下肿瘤,边界清晰,肿瘤常累及椎间孔致椎间孔扩大,典型者呈哑铃状改变,周围骨质多为压迫性骨质吸收;pPNET软组织肿块较大,边界多不清晰,累及椎间孔时多伴周围骨质溶骨性破坏,形成不典型哑铃征。MR动态增强扫描对两者鉴别具有重要价值,PNET早期迅速增强,上升峰极陡,60s左右即达到高峰水平,然后保持平坦,3.5min内未见明显下降曲线;神经源性肿瘤多呈不均匀中度缓慢强化,大部分在8min时尚未出现峰值[15]。
总之,脊柱pPNET非常少见,预后差。对于儿童或青少年患者脊柱肿瘤,密度或信号不均匀,累及椎体骨质和/或椎管,形成椎管内外较大软组织肿块伴不典型哑铃征时应考虑到pPNET的诊断。CT能够准确地观察椎体及附件的骨质破坏、椎间孔扩大、钙化等,MRI可以清楚地显示病灶的范围及肿块与硬膜、神经根、脊髓、马尾、终丝的关系,对手术方案的制定具有重要的临床意义。
猜你喜欢
健康体检与管理(2022年4期)2022-05-13
感染、炎症、修复(2021年1期)2021-07-28
现代临床医学(2021年3期)2021-07-16
世界最新医学信息文摘(2021年12期)2021-06-09
医学食疗与健康(2021年27期)2021-05-13
浙江医学(2020年9期)2020-07-01
中国现代医药杂志(2020年12期)2020-02-06
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年4期)2019-06-18
浙江中西医结合杂志(2019年4期)2019-05-05
