{110}晶面取向Ag3PO4多面体的水热制备及可见光催化活性
2015-11-03 08:53:15刘春梅张国英张许艳艳高东昭
物理化学学报 2015年10期
刘春梅 张国英张 欣 许艳艳 高东昭
(天津师范大学化学学院,天津市功能分子结构与性能重点实验室,无机-有机杂化功能材料化学省部共建教育部重点实验室,天津 300387)
{110}晶面取向Ag3PO4多面体的水热制备及可见光催化活性
刘春梅 张国英*张 欣 许艳艳 高东昭
(天津师范大学化学学院,天津市功能分子结构与性能重点实验室,无机-有机杂化功能材料化学省部共建教育部重点实验室,天津 300387)
采用简易水热法在聚乙二醇-6000 (PEG-6000)辅助下合成了Ag3PO4多面体. 系统考察了水热反应温度、时间及PEG-6000用量对产物形貌和结构的影响. 通过X射线衍射(XRD), 扫描电子显微镜(SEM), 紫外-可见漫反射光谱(UV-Vis DRS)和荧光(PL)光谱等测试手段对光催化剂进行了表征. 结果表明, 适宜的水热温度及PEG-6000用量是制备具有{110}活性晶面取向Ag3PO4多面体的必要条件, 该多面体通过纳米颗粒的Ostwald熟化效应生长而成. 可见光催化降解罗丹明B (RhB)的实验表明, 该Ag3PO4多面体活性明显优于其它水热条件下所制备的非{110}取向晶面样品和离子交换法所得纳米颗粒, 其降解反应速率常数(k)为离子交换法所得Ag3PO4纳米颗粒的8.3倍. 总有机碳含量(TOC)及循环实验证明, 该Ag3PO4多面体可以有效地矿化RhB并保持较好的循环稳定性. 活性自由基捕获实验表明, 空穴(h+)和羟基自由基(OH)是光催化氧化的主要活性物种. 结合活性物种的氧化还原电位以及Ag3PO4的能带结构分析, 提出了催化反应界面光生电子-空穴(e--h+)对的分离及转移机制.
Ag3PO4; PEG-6000; 水热法; 光催化; 活性物种; 光生载流子分离
1 引 言
半导体光催化剂在解决全球能源危机和环境污染等方面具有良好的应用前景.1-5然而迄今为止,半导体材料在很大程度上仍受制于可见光响应范围较窄和光生电子-空穴(e--h+)对复合速率较快等因素,6,7研制和开发可见光响应的新型高效光催化剂一直备受关注.8-10Ag3PO4是近年来新研发的一种可见光响应型光催化剂,11其可见光吸收波长范围在400-560 nm之间,而且电子结构的理论计算表明,Ag3PO4导带的高度离域和的诱导效应均有助于电子-空穴对的分离,12在可见光照射下量子效率可高达92%,表现出极强的氧化能力和分解有机物的能力.
自2010年Ye课题组11在Nature Materials上首次报道该可见光催化剂以来,各种形貌和结构的Ag3PO4相继报道,如零维结构的纳米晶体,13一维结构的纳米纤维、14微米管15和纳米项链,16二维结构的纳米树枝17以及三维结构的四面体、18菱形十二面体19和三棱柱20等. 其中多面体Ag3PO4结构因具有独特的高活性晶面而表现出优越的光催化性能. 如Bi等18采用离子交换法通过H2O2氧化Ag箔合成四面体Ag3PO4结构; Teng等21采用水热法,利用尿素中和磷酸释放NH3实现沉淀-溶解-重结晶的过程,制备出新型四足Ag3PO4结构; Wang等20利用二甲基甲酰胺(DMF)和水作为混合溶剂,通过调节静电作用和超声时间合成三棱柱形Ag3PO4结构. 但是上述多面体Ag3PO4的合成中也均存在不足之处,如使用了价格更为昂贵的Ag箔和有毒的DMF有机溶剂,或者产生了污染性气体NH3等.
然而制备具有高活性取向晶面的Ag3PO4又势在必行,由于异相光催化反应发生在半导体的表面,因此半导体表面的性质和结构对光催化反应的活性和效率具有至关重要的影响. 但是高活性晶面因其具有较高的表面能,通常会在形成过程中快速消失,而表面活性剂或包覆剂可以通过在特定晶面的吸附降低其表面能,从而改变该晶面的生长速度,形成不同形状的纳米晶.22聚乙二醇类作为一种普遍的表面活性剂,价廉无毒且无刺激性,对于制备特定形貌的纳米材料具有关键作用.23
因此基于绿色环保的理念,本文在PEG-6000辅助下采用简易水热法制备出多面体Ag3PO4结构. 通过系统调控水热反应条件,确定了制备具有{110}晶面取向生长Ag3PO4多面体的实验参数,该多面体对染料罗丹明B (RhB)表现出明显优势的可见光催化降解活性,其反应速率常数k是离子交换法所得Ag3PO4纳米颗粒的8.3倍,从而明确了该水热方法中制备-结构-性能之间的关系. 此外,结合活性自由基捕获技术及惰性气氛对比实验,确定了催化反应的主要活性物种; 并进一步结合Ag3PO4的能带结构分析,提出了催化反应界面光生载流子的迁移过程.
2 实验部分
2.1 试剂及实验步骤
AgNO3购于国药集团化学试剂有限公司,异丙醇购于阿拉丁试剂公司,其它试剂购于天津市光复科技发展有限公司,且均为分析纯.
Ag3PO4多面体的制备采用简易水热法. 具体操作如下: 将0.3801 g (1.0 mmol) Na3PO412H2O和0.5100 g (3.0 mmol) AgNO3分别溶于8.0 mL蒸馏水中,向AgNO3溶液中加入0.015 g PEG-6000,搅拌15 min后向其中滴加Na3PO412H2O溶液,继续搅拌30 min后,将所得黄色悬浊液转移至25 mL水热反应釜中并于120 °C反应12 h. 冷却至室温后收集产物,分别用去离子水和乙醇洗涤4-5次,在60 °C空气气氛中干燥4 h,得到黄绿色的Ag3PO4粉末. 调控PEG-6000用量和水热温度等实验参数,得到不同系列Ag3PO4样品.
离子交换法所得Ag3PO4纳米颗粒是在室温下通过AgNO3和Na3PO412H2O反应而得,作为对比实验,编号为RT (室温反应).
2.2 样品表征
采用X射线粉末衍射(XRD)仪(德国布鲁克D8-Advance型)对样品组成和晶型进行分析,选用Cu Kα射线,λ = 0.15418 nm,管电压40 kV,管电流40 mA,扫描范围5°-80°; 采用场发射扫描电子显微镜(FE-SEM,FEI公司NOVA Nano SEM 230)对样品形貌进行分析; 采用附有积分球的紫外-可见分光光度计(日本JASCO公司 V-550/V-570型)测定样品的紫外-可见漫反射光谱(UV-Vis DRS); 采用荧光光谱仪(Hitachi F-4500型)测定样品的荧光光谱; 采用比表面积及孔隙分析仪(美国麦克ASAP2020型)测定样品的比表面积; 采用紫外-可见光谱仪(日本岛津公司UV-2550型)测定RhB溶液的吸收光谱; 采用总有机碳含量(TOC)分析仪(德国Elementar vario)测定RhB溶液的总有机碳含量.
2.3 光催化活性测试
Ag3PO4光催化剂的性能测试在XPA-7型光化学反应仪中完成. 降解液为1 × 10-5molL-1RhB染料溶液,光源为250 W氙灯附420 nm滤光片. 具体操作如下: 取10.0 mL RhB溶液于石英试管中,加入5.0 mg Ag3PO4光催化剂,将该悬浊液在暗箱中磁力搅拌30 min以建立吸附-脱附平衡. 开始光照后,每间隔5 min取出一支石英试管,立即离心分离去除光催化剂,利用紫外-可见分光光度计检测上清液的吸光度,以计算降解效率.
3 结果与讨论
3.1 组成及晶相
图1是离子交换法和不同PEG-6000用量水热法所得Ag3PO4样品的XRD图谱. 所有样品的各衍射峰位置均与体心立方结构的Ag3PO4(JCPDS No. 06-0505)吻合,未检测到Ag2O和单质Ag杂相的XRD峰,说明合成了高纯度的Ag3PO4样品. 但值得关注的是,PEG-6000用量在0.010-0.020 g范围内的样品具有明显的{110}晶面生长取向,其中(110)/(200)的衍射峰强度比分别为1.14、1.71、1.51 (图1(c-e)),而标准卡片和离子交换样品(图1a)的相应比值仅为0.77和0.91. Ye等19也曾在菱形十二面体中报道过类似的情况,并证明{110}晶面比{100}晶面具有更高的表面能和更丰富的活性位点. 而无PEG-6000 (图1b)及其用量为0.030 g (图1f)时,水热样品的(110)/(200)峰强度比分别为0.84和0.72. 显然,PEG-6000用量可调控{110}晶面的暴露比例,且随其用量的增加呈现强度先增后减的趋势.
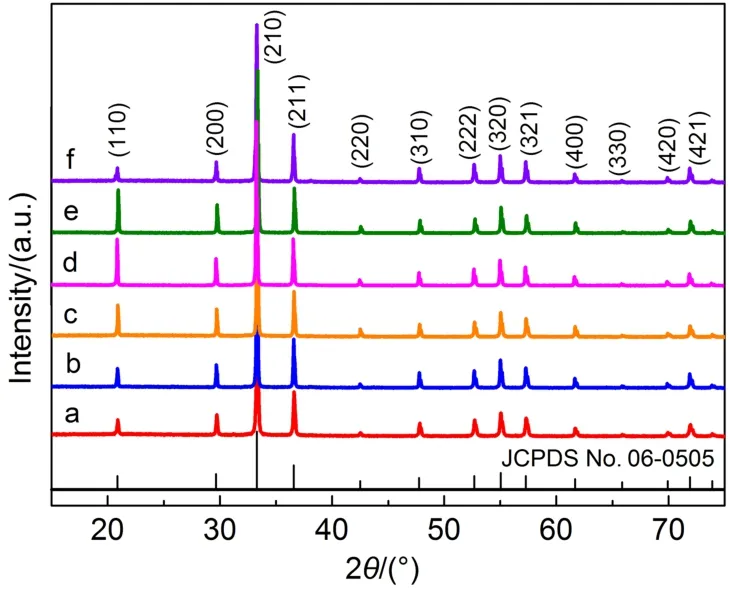
图1 离子交换法及水热所得系列Ag3PO4样品的XRD图Fig.1 XRD patterns of the Ag3PO4samples prepared by ion-exchange method and hydrothermal reaction
3.2 制备参数的影响
3.2.1 PEG-6000用量的影响
图2是不同PEG-6000用量所得Ag3PO4样品的SEM图,产物的基本形貌为粒径约2-4 µm的多面体,但PEG-6000的用量对于多面体的完整性有明显影响. 如图2a所示,当水热体系中不存在PEG-6000时,所得样品的形貌以不规则多面体和近球状微粒为主,且表面多存在如插图所示的孔洞. 随着PEG-6000用量的增加,球状微粒有所减少,尤其当该表面活性剂用量为0.015 g时(图2c),多面体逐渐趋于完整,呈现出更加清晰的棱和角. 然而进一步增加PEG-6000用量(图2(d,e)),产物中近球状颗粒又明显增加,且多面体棱角的清晰度减弱. 这说明适量PEG-6000的辅助有利于较规则多面体的形成,过量表面活性剂对晶核的包裹反而抑制了Ag3PO4小颗粒Ostwald熟化过程.
3.2.2 水热反应温度的影响
图3是不同水热温度所制备Ag3PO4样品的SEM及XRD图谱. 显然水热温度对产物的组成、晶相结构及形貌均有明显影响. 在80 °C的低温水热条件下,样品中大量Ag3PO4纳米颗粒和少数不规则多面体共存,说明低温水热不利于纳米颗粒的溶解-重结晶过程. 随着水热温度的升高,纳米颗粒逐渐减少而多面体明显增多. 当温度升至120 °C,纳米颗粒已基本消失,Ag3PO4多面体生长较完整且棱角清晰.相应的XRD图谱则反映出{110}晶面有明显取向生长,其中120 °C时(110)晶面的衍射峰强度比例最高,其强度是80 °C时的3倍. 然而进一步升温时,多面体及棱角的清晰度开始降低,并伴随XRD中{110}晶面的优势取向减弱. 尤其是160 °C样品已基本转变为球体结构,(110)晶面不但失去了优势取向,而且产物中出现了少量Ag (JCPDS No. 04-0783)杂质. 因此,适宜的水热温度也可调控具有最强{110}晶面取向Ag3PO4多面体的形成.

图2 不同PEG-6000用量所得Ag3PO4样品的SEM图Fig.2 SEM images of Ag3PO4samples with different dosages of PEG-6000
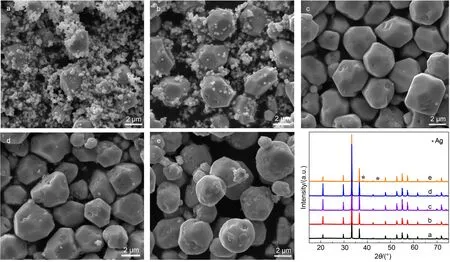
图3 不同水热温度所制备Ag3PO4样品的SEM图及相应的XRD图谱Fig.3 SEM images and the corresponding XRD patterns of Ag3PO4samples prepared at different hydrothermal temperatures
3.2.3 水热反应时间的影响
控制反应温度,研究不同反应时间所得产物的形貌有助于了解Ag3PO4多面体的生长过程. 图4为120 °C不同水热时间所得Ag3PO4样品的SEM图. 如图4a所示,未经水热处理的前驱体即离子交换法所得样品为粒径100-200 nm的小颗粒. 水热反应40 min时(图4b),纳米颗粒中出现少量约1-2 µm的多面体. 延长水热反应时间至2 h,样品中的多面体明显增多,而纳米颗粒大大减少. 随着水热反应时间的继续增加,纳米颗粒逐渐消失而Ag3PO4多面体趋于完整. 尤其水热反应12 h时,出现完整、均匀、棱角清晰的多面体. 以上演化过程表明,多面体Ag3PO4的形成所遵循的是典型的Ostwald熟化过程. 根据吉布斯-汤姆森公式:24ln(c/cs) = 2σM/dRTr (其中c为溶液相浓度(molL-1),cs为溶液的饱和浓度(molL-1),σ为表面张力(Nm-1),M为晶体分子的摩尔质量(gmol-1),d为晶体密度(kgm-3),R为摩尔气体常数(Jmol-1K-1),T为绝对温度(K),r为临界晶核半径(m)),晶核的半径r越小则其溶解度越大,因此随着水热反应时间的延长,Ag3PO4纳米小颗粒逐渐溶解并重新结晶在较大颗粒表面. 而PEG-6000在该多面体形成过程中起着不可或缺的作用. 一方面,作为一种有机封端机,其亲水性的―CH2―CH2―O―易与Ag+形成Ag-PEG配位键,有效减少了溶液中Ag+的浓度,因此可减缓Ag3PO4晶核的形成速度,有利于晶核的生长;25而在晶核长大的过程中,PEG-6000则起到了选择吸附和诱导结晶作用,根据文献19密度泛函理论的计算结果,{110}比{100}具有更高的表面能和活性,因此富含羟基的PEG-6000将更易优先吸附于{110}晶面,并在其辅助引导下使Ag+和沿该晶面继续结晶,最终形成较为完整的{110}晶面取向Ag3PO4多面体.

图4 120 °C时不同水热时间所得Ag3PO4样品的SEM图Fig.4 SEM images of Ag3PO4samples treated at 120 °C with different hydrothermal time
3.3 光学性质
图5a为不同PEG-6000用量所得Ag3PO4样品的紫外-可见吸收光谱. 从图中可以看出Ag3PO4的吸收阈值位于可见光区域,陡而直的峰形表明该吸收来自Ag3PO4的本征带隙跃迁,而非存在杂质跃迁. 相比不加表面活性剂的样品,PEG-6000用量为0.015g时所得样品对太阳光的利用率更高,吸收带边发生约20 nm的红移. 该现象可能是由于PEG辅助样品中基本不存在Ag3PO4纳米颗粒,因而不会出现量子尺寸效应所导致的吸光蓝移效应所致. 根据光电效应公式αhv = A(hv - Eg)n/2可计算带隙能,其中α、h、v、Eg、A分别为吸光系数(cm-1)、普朗克常数(Js-1)、频率(s-1)、带隙能(eV)和常数,而n决定半导体的跃迁类型,当n = 1时为直接跃迁,而当n = 4时为间接跃迁. 由于Ag3PO4为间接跃迁半导体,12如插图所示以(αhv)1/2对hv作图获得多面体Ag3PO4的带隙宽度为2.18 eV,相应于569 nm的吸收带边,对太阳光中的可见光有较高利用率.
荧光光谱常用来考察半导体中光生电子-空穴对的分离与复合,荧光强度的降低常意味着光生载流子的复合几率被抑制.26图5b是在350 nm紫外光激发下不同PEG-6000用量所得Ag3PO4的荧光光谱,所有样品的荧光峰形基本保持一致,551 nm的最强荧光峰归属于激发电子从导带跃迁回价带,从而引起光生载流子复合而释放的能量. 但当PEG-6000用量为0.015 g时,样品的荧光强度最低,说明该样品中光生载流子的复合几率最小,有利于光量子效率及光催化活性的提高. 根据图1的 XRD分析结果,该条件下所制备的Ag3PO4多面体具有{110}晶面取向生长,因其具有高的表面能和丰富的活性位点,19因此光生载流子可以得到更为快速的分离与迁移.
3.4 光催化性能
以RhB染料为降解探针,系统评价了系列Ag3PO4样品的光催化活性. 空白对照实验表明染料吸附及无催化剂存在下的光解效应极其微弱,因此均可忽略不计. 图6a为各样品对RhB的光催化降解效率对比,离子交换法所得Ag3PO4纳米颗粒的光催化活性最低,光辐照30 min后对染料的降解效率仅为30.5%. 所有水热样品均具有明显提高的光催化活性,但不同水热参数对样品的光催化性能有明显影响,其中PEG-6000用量为0.015 g,水热温度为120 °C时所制备的Ag3PO4样品光催化性能最佳,同等条件下对RhB分子的降解率可高达97.5%. 由前面对不同条件所制备样品的XRD及SEM分析可知,该条件下Ag3PO4多面体生长最完整,且具有最强{110}晶向取向生长,而据文献19报道{110}晶面是Ag3PO4的高活性晶面(1.31 Jm-2),其比{100}晶面(1.12 Jm-2)具有更高的表面能,并且具有更丰富的氧空位. 因此该晶面不但有利于O2吸附和光生电子转移过程的进行,而且有利于有机污染物在其表面的吸附及氧化降解反应的进行,从而表现出最高的光催化活性.而其它条件如PEG-6000用量为0.010 g的所得Ag3PO4,虽然催化活性也有显著提高,但由图1可知此时样品的{110}取向晶面尚未达到最高比例,因此其光降解效率也并非最佳. 该结果进一步证实了制备参数、材料结构以及使用性能三者之间的内在影响.

图5 Ag3PO4光催化剂的紫外-可见漫反射光谱(UV-Vis DRS)和荧光(PL)光谱(λex= 350 nm)Fig.5 UV-Vis diffuse reflectance spectra (UV-Vis DRS) and photoluminescence (PL) (λex= 350 nm)spectra of Ag3PO4photocalalyst
对于低浓度染料而言,其降解反应符合准一级反应动力学,27图6a中系列Ag3PO4样品相应的一级催化降解动力学曲线如图6b所示,均具有较好的线性相关系数. 其中生长完整Ag3PO4多面体的反应速率常数k = 0.10026 min-1,为无PEG-6000辅助所得水热Ag3PO4样品(k = 0.01560 min-1)的6.4倍,而达到离子交换法所得Ag3PO4纳米颗粒(k = 0.01204 min-1)的8.3倍. 比表面测试表明该Ag3PO4多面体的BET面积为0.85 m2g-1,仅为离子交换法所得样品(6.57 m2g-1)的13%,但却表现出显著优势的光催化活性,进一步说明高活性{110}取向晶面在该Ag3PO4多面体光催化中起着主要作用. 为了更清楚对比不同Ag3PO4样品对RhB的催化降解过程,图6(c,d)分别给出了在无PEG-6000辅助及其用量为0.015 g时所得Ag3PO4催化下RhB的吸收光谱变化. 整个脱色过程中RhB最大吸收峰并未出现从553 nm向498 nm蓝移的现象,说明该过程中不存在RhB的光敏化作用,降解主要来自Ag3PO4的光催化作用.28同等条件下,具有{110}晶面取向生长的Ag3PO4多面体光催化反应30 min即可基本使RhB脱色完全,而无PEG-6000辅助所得水热样品对RhB的降解率仅为38%,进一步直观显示了该多面体优异的光催化活性.
染料溶液的降解脱色并不能代表污染物中有机物质的彻底矿化. 为了进一步研究所制备{110}晶面取向Ag3PO4多面体对RhB的矿化效率,测定了其在不同辐照时间的TOC去除率,并与无PEG-6000辅助所得样品进行了对比. 如图7a所示,在Ag3PO4多面体的催化下,随着光照时间的延长,RhB染料溶液逐渐被矿化为无机小分子,60 min后对RhB的矿化率达到42%. 而对于非明显{110}晶面取向的Ag3PO4样品,30 min后矿化程度缓慢,60 min时的TOC去除率仅为17%,低于Ag3PO4多面体约25%. 该结果进一步说明{110}晶面在Ag3PO4多面体光催化降解和矿化中的高活性作用.
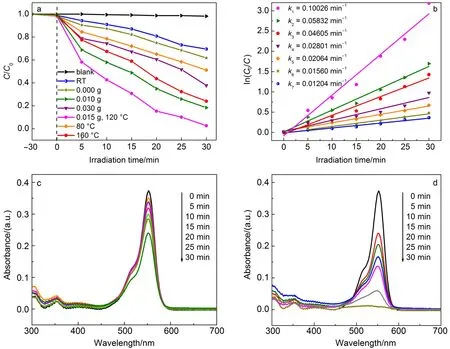
图6 (a)不同Ag3PO4样品光催化降解RhB的活性,(b)相应的一级反应动力学曲线及速率常数(k),(c)无PEG-6000及(d)其用量为0.015 g时Ag3PO4水热样品对RhB的催化降解过程Fig.6 (a) Degradation of RhB over series of Ag3PO4samples,(b) the corresponding first-order dynamic plots and rate constant k; photocatalytic degradation processes of RhB over Ag3PO4hydrothermal samples obtained (c) without and(d) with 0.015 g PEG-6000
光催化剂良好的循环稳定性是影响其应用前景的重要因素. 该Ag3PO4多面体由于具有微米尺寸,仅通过简单过滤甚至自然沉降即可回收,从而可避免对环境的二次污染. 从图7b可知,该{110}晶面取向Ag3PO4多面体在前3次循环中保持了良好的光催化活性,虽然第4次循环的催化活性有所降低,但光照40 min后仍能保持约80%的降解率. 然而非{110}晶面取向Ag3PO4样品的催化活性随循环次数的增加明显降低,第4次循环后其对RhB的降解率仅为第1次循环的42%. 众所周知,Ag3PO4因光腐蚀4,29而稳定性不够理想,但显然该{110}晶面取向对Ag3PO4的稳定性具有明显正面效应. 这应该是由于其高的表面能和丰富的氧空位均有利于光生电子的快速转移,从而抑制了Ag+因捕获电子而被还原的过程.
3.5 光催化机理研究
为了研究在Ag3PO4多面体催化下,氧化降解RhB染料分子的主要活性物种,分别进行了空穴、自由基捕获剂存在下以及惰性气氛下的光降解对比实验. 如图8所示,非捕获剂或惰性气氛中,30 min辐照后Ag3PO4多面体对RhB的降解率高达97%.而当体系中加入空穴(h+)捕获剂乙二胺四乙酸(EDTA)后,降解效率大幅降低,同等条件下仅有15%的RhB得到脱色; 羟基自由基(OH)捕获剂异丙醇IPA的加入同样严重抑制了光催化效果,其降解效率也只有约18%. 该实验结果说明h+和OH均是光催化反应中的主要活性物种. 此外,若光生电子被催化剂表面的吸附O2捕获,可生成超氧自由基它往往也是光催化反应的活性物种之一. 为了验证在该催化过程中的作用,进行了惰性气氛下的光降解对比实验,显然无氧条件下RhB的光降解效率只有微弱降低,说明并不是该催化过程的主要活性物种.
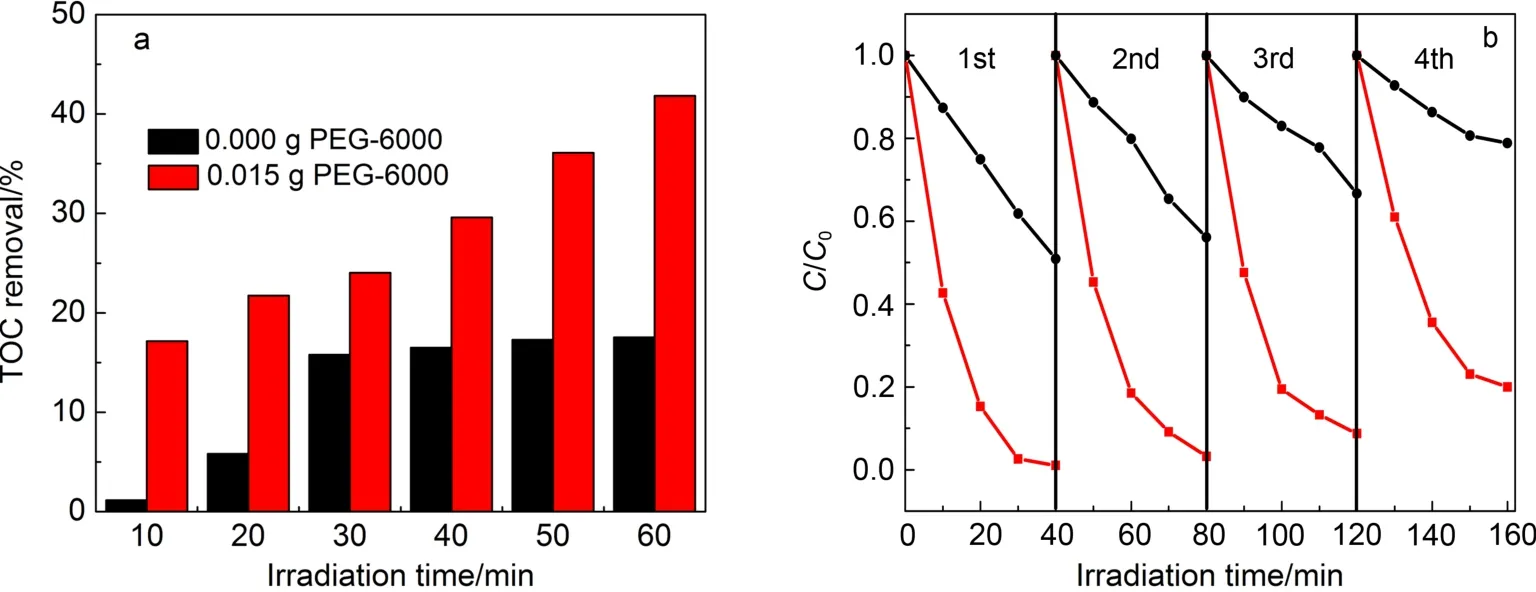
图7 无PEG-6000及其用量为0.015 g时Ag3PO4样品对RhB的(a) TOC分析和(b)循环性能对比Fig.7 Comparison of (a) TOC analysis and (b) circulation runs for RhB over Ag3PO4samples obtained without and with 0.015 g PEG-6000
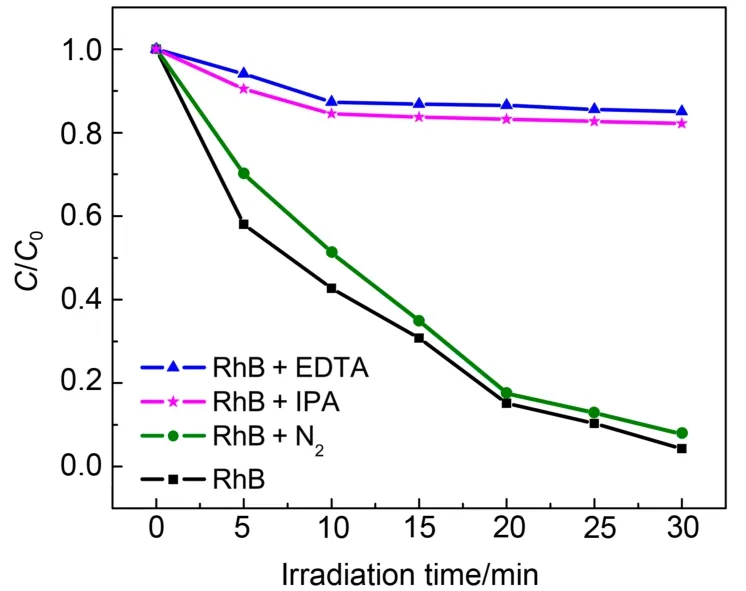
图8 捕获剂对Ag3PO4光催化降解RhB的影响Fig.8 Effects of different scavengers on the degradation of RhB in the presence of Ag3PO4photocatalysts
活性物种的氧化还原电位与半导体价带(VB)导带(CB)位置间的关系是决定光生载流子分离与迁移的决定性因素. 半导体光催化剂的VB和CB电势可以通过元素的绝对电负性公式 ECB= X - Ec-1/2Eg进行理论预测. 其中,X是半导体的绝对电负性,其数值为半导体各元素绝对电负性的几何平均值; Ec为相对于氢电极电位的自由电子能量(约4.5 eV); Eg是半导体的禁带宽度. 根据DRS光谱所测带隙能2.18 eV,通过上式计算Ag3PO4的VB和CB电位分别为2.55和0.37 V,与文献30报道吻合. 结合该电位值与前面活性物种的分析结果,提出如图9所示的Ag3PO4表面光生载流子迁移机理. 在可见光激发下,Ag3PO4多面体中分别产生导带e-和价带h+. 该h+较正的电位(2.55 V)表明其具有强氧化性,而实验也已证实它是光催化反应的的主要活性物种之一,因此h+可直接氧化吸附于Ag3PO4表面的有机污染物. 而另一活性物种OH则可以通过h+或e-两种转移途径分别产生. 从价带h+的角度来讲,由于其电位比OH/OH-(H2O)31的标准电极电势值2.38 V更正,因此可将降解液中的OH-或H2O氧化为OH; 从导带e-的角度而言,其可被吸附于催化剂表面的O2分子所捕获,32但由于的单电子转移电极电势为-0.046 V (vs SHE(标准氢电极)),33比Ag3PO4的导带电位(0.37 V)明显更负,因此该单电子转移为热力学禁阻过程,即体系中不易产生与前面所分析并非活性物种结果一致. 但O2捕获双电子反应O2+ 2e-+ 2H+= H2O2(aq)的标准电极电势为+0.682 V (vs SHE),34比Ag3PO4的导带电位更正,因此O2可通过该双电子转移过程还原为H2O2,并进一步分解为OH. 通过上述过程,Ag3PO4中的光生e--h+对得到了有效分离,并一步转化为活性氧化物种,参与有机污染物的光催化降解过程.

图9 Ag3PO4光催化剂表面光生载流子分离及迁移示意图Fig.9 Schematic illustration of the charge separation and immigration on the surface of Ag3PO4photocatalyst
4 结 论
通过简易水热法合成了具有{110}晶面取向生长的Ag3PO4多面体,参数探讨实验表明PEG-6000用量和水热温度是制备该特征多面体的重要影响因素. 相比于离子交换法所得Ag3PO4纳米颗粒,以及其它水热条件下所得不具备晶面取向生长的颗粒,该多面体表现出明显优异的可见光光催化活性. 该研究内容丰富了材料的制备参数、结构特点及使用性能间的相互影响关系. 在此基础上,进一步深入探讨了Ag3PO4多面体光催化氧化的主要活性物种,并提出了该光催化剂反应界面光生电子-空穴的分离与迁移途径.
(1)Fujishima,A.; Honda,K. Nature 1972,238,37. doi: 10.1038/238037a0
(2)Mohamed,S. H.; El-Hagary,M.; Althoyaib,S. Eur. Phys. J. -Appl. Phys. 2012,57,20301. doi: 10.1051/epjap/2012110312
(3)Wang,X. X.; Xu,H. L.; Shen,W.; Ruhlmann,L.; Qin,F.;Sorgues,S.; Colbeau-Justin,C. Acta Phys. -Chim. Sin. 2013,29,1837. [王晓夏,徐华龙,沈 伟,Ruhlmann L.,秦 枫,Sorgues S.,Colbeau-Justin C. 物理化学学报,2013,29,1837.] doi: 10.3866/PKU.WHXB201307024
(4)Ge,M.; Tan,M. M.; Cui,G. H. Acta Phys. -Chim. Sin. 2014,30,2107. [葛 明,谭勉勉,崔广华. 物理化学学报,2014,30,2107.] doi: 10.3866/PKU.WHXB 201409041
(5)Nakamura,K. J.; Ide,Y.; Ogawa,M. Mater. Lett. 2011,65,24. doi: 10.1016/j.matlet.2010.09.043
(6)Tian,G. H.; Fu,H. G.; Jing,L. Q.; Xin,B. F.; Pan,K. J. Phys. Chem. C 2008,112,3083. doi: 10.1021/jp710283p
(7)Chen,X. B.; Shen,S. H.; Guo,L. J.; Mao,S. S. Chem. Rev. 2010,110,6503. doi: 10.1021/cr1001645
(8)Cheng,H. F.; Huang,B. B.; Dai,Y.; Qin,X. Y.; Zhang,X. Y. Langmuir 2010,26,6618. doi: 10.1021/la903943s
(9)Wang,D. F.; Kako,T.; Ye,J. H. J. Am. Chem. Soc. 2008,130,2724. doi: 10.1021/ ja710805x
(10)Yuhas,B. D.; Smeigh,A. L.; Douvalis,A. P.; Wasielewski,M. R.; Kanatzidis,M. G. J. Am. Chem. Soc. 2012,134,10353. doi: 10.1021/ja303640s
(11)Yi,Z. G.; Ye,J. H.; Kikugawa,N.; Kako,T.; Ouyang,S. X.;Stuart-Williams,H. Nat. Mater. 2010,9,559. doi: 10.1038/NMAT2780
(12)Ma,X. G.; Lu,B.; Li,D.; Shi,R.; Pan,C. S.; Zhu,Y. F. J. Phys. Chem. C 2011,115,4680. doi: 10.1021/jp111167u
(13)Dinh,C. T.; Nguyen,T. D.; Kleitz,F.; Do,T. O. Chem. Commun. 2011,47,7797. doi: 10.1039/c1cc12014j
(14)Yu,H. C.; Dong,Q. S.; Jiao,Z. B.; Wang,T.; Ma,J. T.; Lu,G. X.; Bi,Y. P. J. Mater. Chem. A 2014,2,1668. doi: 10.1039/c3ta14447j
(15)Hua,X.; Jin,Y. J.; Wang,K.; Li,N.; Liu,H. Q.; Chen,M. D.;Paul,S. S.; Zhang,Y.; Zhao,X. D.; Teng,F. Catal. Commun. 2014,52,49. doi.org/10.1016/j
(16)Bi,Y. P.; Hu,H. Y.; Ouyang,S. X.; Jiao,Z. B.; Lu,G. X.; Ye,J. H. J. Mater. Chem. 2012,22,14847. doi: 10.1039/c2jm32800c
(17)Bi,Y. P.; Hu,H. Y.; Jiao,Z. B.; Yu,H. C.; Lu,G. X.; Ye,J. H. Phys. Chem. Chem. Phys. 2012,14,14486. doi: 10.1039/ c2cp42822a
(18)Hu,H. Y.; Jiao,Z. B.; Yu,H. C.; Lu,G. X.; Ye,J. H.; Bi,Y. P. J. Mater. Chem. A 2013,1,2387. doi: 10.1039/c2ta01151d
(19)Bi,Y. P.; Ouyang,S. X.; Umezawa,N.; Cao,J. Y.; Ye,J. H. J. Am. Chem. Soc. 2011,133,6490. doi: org/10.1021/ja2002132
(20)Dong,P. Y.; Wang,Y. H.; Li,H. H.; Li,H.; Ma,X. L.; Han,L. L. J. Mater. Chem. A 2013,1,4651. doi: 10.1039/c3ta00130j
(21)Wang,J.; Teng,F.; Chen,M. D.; Xu,J. J.; Song,Y. Q.; Zhou,X. L. CrystEngComm 2013,15,39. doi: 10.1039/c2ce26060c
(22)Yin,Y. D.; Alivisatos,A. P. Nature 2005,437,664. doi: 10.1038/nature04165
(23)Hu,L. M.; Lin,C. G.; Wang,L.; Yuan,S. L. Acta Phys. -Chim. Sin. 2014,30,2149. [胡立梅,蔺存国,王 利,苑世领. 物理化学学报,2014,30,2149.] doi: 10.3866/PKU.WHXB201409021
(24)Mullin,J. W.; Yokota,M.; Mullin,J. W. J. Cryst. Growth 1997,182,86. doi: 10.1016/S0022-0248(97)00328-X
(25)Hua,X.; Jin,Y. J.; Wang,K.; Li,N.; Liu,H. Q.; Chen,M. D.;Paul,S. S.; Zhang,Y.; Zhao,X. D.; Teng,F. Catal. Commun. 2014,52,49. doi: 10.1016/j.catcom.2014.04.014
(26)Cui,G. W.; Wang,W. L.; Ma,M. Y.; Zhang,M.; Xia,X. Y.;Han,F. Y.; Shi,X. Y.; Zhao,Y. Q.; Dong,Y. B.; Tang,B. Chem. Commun. 2013,49,6415. doi: 10.103/c3cc42500b
(27)Zhang,C.; Zhu,Y. F. Chem. Mater. 2005,17,3537. doi: 10.1021/cm0501517
(28)Wu,T. X.; Liu,G. M.; Zhao,J. C.; Hiodaka,H.; Serpone,N. J. Phys. Chem. B 1998,102,5845. doi: 10.1021/jp980922c
(29)Smith,W.; Mao,S.; Lu,G. H.; Catlett,A.; Chen,J. H.; Zhao,Y. P. Chem. Phys. Lett. 2010,485,171. doi: 10.1016/j.cplett. 2009.12.041
(30)Indra,A.; Menezes,P. W.; Schwarze,M.; Driess,M. New J. Chem. 2014,38,1942. doi: 10.1039/c3nj01012k
(31)Cheng,H. F.; Huang,B. B.; Dai,Y.; Qin,X. Y.; Zhang,X. Y. Langmuir 2010,26,6618. doi: 10.1021/la903943s
(32)Ma,S. S.; Li,R.; Lv,C. P.; Xu,W.; Gou,X. L. J. Hazard. Mater. 2011,192,730. doi: 10.1016/j.jhazmat.2011.05.082
(33)Ye,L. Q.; Chen,J. N.; Tian,L. H.; Liu,J. Y.; Peng,T. Y.; Deng,K. J.; Zan,L. Appl. Catal. B 2013,130-131,1. doi: 10.1016/ j.apcatb.2012.10.011
(34)Liu,W.; Wang,M. L.; Xu,C. X.; Chen,S. F.; Fu,X. L. Mater. Res. Bull. 2013,48,106. doi: 10.1016/j.materresbull.2012.10.015
摘要: 三种多级结构花状硫化铜通过水热法, 利用纳米薄片自组装形成. 加入有机分子聚乙烯吡咯烷酮或1,3,5-均苯三甲酸调控其片层密度. 产物作为基板生长镍纳米颗粒. 通过环境扫描电子显微镜(SEM), X射线衍射(XRD), 透射电子显微镜(TEM)等对这种复合物的结构进行表征. 利用紫外-可见吸收光谱, 研究了产物作为催化剂催化还原4-硝基苯酚的性能. 结果表明, 长在具有最稀疏片层的样品(Ni@SUB2)上的镍纳米颗粒(Ni NPs, 直径5 nm左右)具有超低负载量, 为0.469% (w). Ni@SUB2在三种Ni@SUB复合物中具有最好的催化性能. 还原4-硝基苯酚的反应中, 4-硝基酚初始浓度为0.2 mmolL-1时Ni@SUB2在4 min中内转化率可以实现100%, 而等量的纯Ni纳米颗粒转化率只有43%. 增强的催化性能可以归结为镍纳米颗粒在硫化铜基板上得到良好分散, 可以提供更多催化位点. 同时硫化铜基板不溶于水, 可以通过离心的方式回收催化剂, 有利于环境保护.
关键词: 基板; 硫化铜; 镍; 纳米催化剂; 4-硝基苯酚
中图分类号: O643
doi: 10.3866/PKU.WHXB201509091
Hydrothermal Synthesis of Ag3PO4Polyhedrons with Oriented {110}Facets and Visible-Light-Driven Photocatalytic Activity
LIU Chun-Mei ZHANG Guo-Ying*ZHANG Xin XU Yan-Yan GAO Dong-Zhao
(Tianjin Key Laboratory of Structure and Performance for Functional Molecules,Key Laboratory of Inorganic-Organic Hybrid Functional Material Chemistry,Ministry of Education,College of Chemistry,Tianjin Normal University,Tianjin 300387,P. R. China)
Ag3PO4polyhedrons were synthesized by a facile hydrothermal route using polyethylene glycol-6000 (PEG-6000). The effects of hydrothermal temperature, reaction time, and PEG-6000 dosage on the morphologies and structures of the products were systematically investigated. The photocatalysts were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), ultraviolet-visible diffuse reflectance spectra (UV-Vis DRS), and photoluminescence (PL) spectra. The hydrothermal temperature and the PEG dosage are key factors in the production of Ag3PO4polyhedrons with oriented {110} facets. The Ag3PO4polyhedrons evolve via Ostwald ripening, and exhibit superior visible-light photocatalytic degradation of Rhodamine B (RhB) relative to Ag3PO4samples without oriented {110} facets and Ag3PO4nanoparticles prepared by anion-exchange. The reaction rate constant of the Ag3PO4polyhedrons was 8.3 times that of the Ag3PO4nanoparticles. Total organic carbon (TOC) analysis and cycling experiments revealed that the polyhedrons have better mineralization efficiency and exhibit good circulation runs. Holes (h+) and hydroxylradicals (•OH) are confirmed to be the dominant active species in the presence of radical scavengers and in N2-saturated solution. Given the redox potential of the active species and the band structure of Ag3PO4polyhedron, the separation and migration mechanism of photogenerated electron-hole (e--h+) pairs at the photocatalytic interface was proposed.
Silver phosphate; PEG-6000; Hydrothermal route; Photocatalysis; Active species;Separation of photogenerated carriers
多级结构Ni@CuS复合物的合成及在提高催化还原4-硝基苯酚性能方面的应用
马亦然 周 苇*曹 薇 郑金龙 郭 林
(北京航空航天大学化学与环境学院,北京 100191)
July 6,2015; Revised: August 21,2015; Published on Web: August 25,2015.
. Email: hxxyzgy@mail.tjnu.edu.cn; Tel: +86-22-23766515.
O643; O644
10.3866/PKU.WHXB201508251
The project was supported by the National Natural Science Foundation of China (21303122) and Program for Innovative Research Team in University of Tianjin,China (TD12-5038).
国家自然科学基金(21303122)和天津市高等学校创新团队培养计划(TD12-5038)资助项目
猜你喜欢
火炸药学报(2022年5期)2022-11-04 02:30:48
数学大王·低年级(2022年3期)2022-03-17 21:51:44
课外生活·趣知识(2021年8期)2021-08-24 02:25:39
物理实验(2019年7期)2019-08-06 05:35:56
航空材料学报(2019年2期)2019-04-15 01:04:08
物理学报(2018年22期)2018-12-18 05:58:28
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:35:26
科学大众·小诺贝尔(2016年6期)2016-08-17 09:43:30
金色年华(2016年11期)2016-02-28 01:42:38
石油化工应用(2014年2期)2014-03-11 17:39:00