
家养动物基因组拷贝数变异研究进展及其育种应用展望
2015-10-28 02:52程红姜雨
生物技术通报 2015年11期
程红 姜雨
(西北农林科技大学 动物科技学院,杨凌 712100)
家养动物基因组拷贝数变异研究进展及其育种应用展望
程红 姜雨
(西北农林科技大学 动物科技学院,杨凌 712100)
家养动物参考基因组组装的不断完善和群体重测序数据的持续增加促进了基因组中大量变异的发现。基因组上的变异主要包括单核苷酸变异(SNP)和拷贝数变异(CNV)两种类型。相对于数量众多,已经被广泛研究和用作分子育种标记SNP,目前已经被发现和经过实验验证其功能的CNV数量较少,鲜有被直接用作分子标记进行育种的报道。CNV片段长度大、在基因组中普遍存在且比SNP变异覆盖的基因组范围更广,所以可能对农艺性状造成很大影响,其在畜禽基因组研究和育种应用中具有广阔前景。重点讨论了家养动物CNV的研究进展,并对其在家养动物育种中的应用进行了分析展望。
家养动物;基因组;拷贝数变异;育种;经济性状
自2004年第一个畜禽基因组——红原鸡[1]全基因组测序完成后,牛[2]、猪[3]、山羊[4]、鸭[5]、绵羊[6]和草鱼[7]等广布性家养动物的参考基因组构建工作相继完成。在此基础上,大量群体重测序研究筛选到数量巨大的基因组变异。
基因组变异分为两类,一类是由单核苷酸替换造成的变异,即我们所熟知的单核苷酸多态性(Single nucleotide polymorphism,SNP),也可称为单核苷酸变异(Single nucleotide variant,SNV);另一类是核苷酸序列上的结构变异(Structure variation,SV)。其中SV又可以分为两种,一种是序列位置变化引起的倒位(Inversion)或易位(Translocation);另一种是由于序列复制或者丢失导致的小于50 bp的小片段插入/丢失(Insertion/Deletion,InDel)和大于50 bp的长片段拷贝数变异(Copy number variation,CNV)[8]。 这些数量巨大的基因组变异是否会影响表型、作用机制是什么,目前的研究远未解决这些问题。而家养动物基因组学研究的核心内容就是解释表型发生的遗传基础,特别是家养动物的不同品种或个体间重要经济性状差异对应的遗传变异机制,这要求我们必须在全基因组水平对变异进行归类分析,解析变异与表型的一一对应关系,最终用于分子育种。
基因组变异中数量最多的是SNP,其也一直是科学家们研究的焦点。2010年,Rubin等[9]在鸡驯化研究中通过重测序发现了超过 700万个SNPs。2014年,Daetwyler等[10]在234头牛重测序研究中鉴定出2 830万个变异,其中2 670万个变异为SNPs。遍布于整个基因组SNP现已作为主要的分子标记用于辅助育种。但是,单个SNP仅造成了一个核苷酸的改变,可能只有极少数最终能够影响基因表达或改变蛋白编码序列,进而造成表型的改变。2014年,Carneiro等[11]在家兔驯化过程的遗传改变研究中证实了这一问题。他们检查了从野兔到家兔的所有SNPs,共找到5 000万个SNPs 。其中只有7万个SNPs造成了氨基酸编码的错义突变,而在驯化过程中固定下来的错义突变SNPs只有14个,没有发现在驯化中固定下来的引起无义或移码突变的SNP,而少数被固定的SNPs大都不在编码位点和非编码的保守位点。该研究并没有探讨CNV对驯化的影响。
相对于SNP,CNV属于大片段的结构变异,能够影响一个较宽区域的基因组序列,甚至改变一些基因的序列、剪切和表达。由于检测手段变得更精细,2014年,MacDonald等[8]对CNV的长度进行了重新定义,将CNV长度从以往的基因组中1 000 bp以上的重复或缺失改为50 bp以上的重复或缺失,并以此作为长片段拷贝数变异的长度阈值。目前,CNV在人类及许多模式生物物中得到了广泛的研究(表1),家养动物的拷贝数变异研究也在陆续展开。CNV已被认为是一种重要的遗传变异来源,与重要经济性状以及物种进化过程密切相关,将作为新的分子遗传标记在表型多态性和进化过程研究中扮演重要角色。
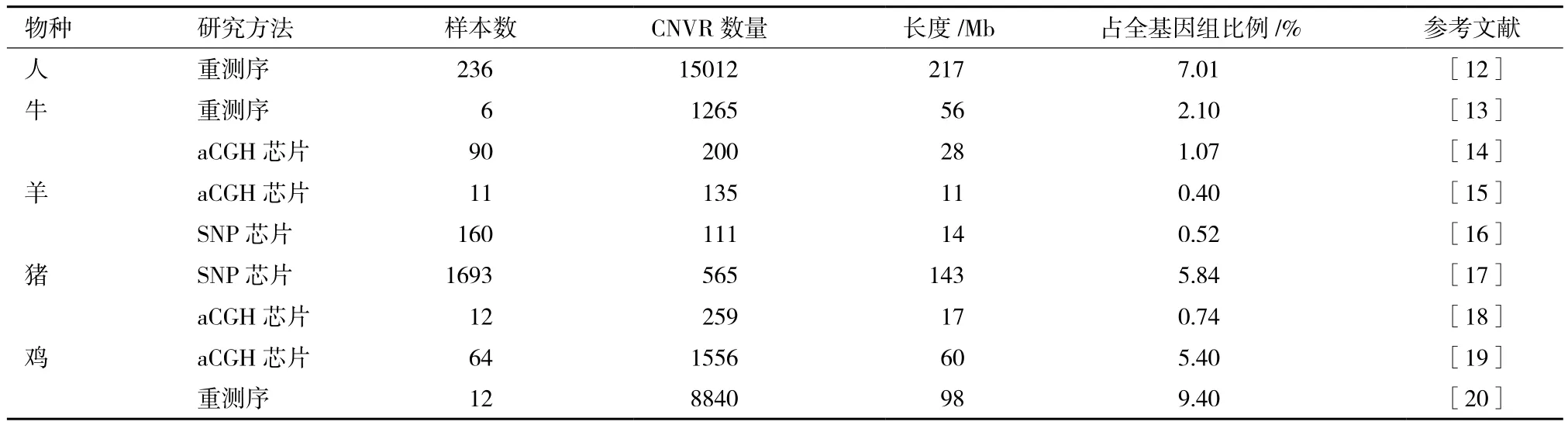
表1 人和家养动物CNV区域研究比较
1 CNV是广泛存在的重要遗传变异
随着基于微阵列的比较基因组杂交技术(arraybased comparative genomic hybridization,aCGH)和第二代高通量测序技术的快速发展,基因组更大范围及更多数量的CNV变异被发现。
2004年,Sebat等[21]发现CNV广泛存在于人类基因组中,鉴定出221个大于100 kb的CNVs。2006年,Redon等[22]构建了人类基因组首个 CNV图谱,共鉴定得到1 447个拷贝数变异区域(Copy number variation regions,CNVRs),覆盖的核苷酸总数达到360 Mb,远远超过了人类SNP总数,其中近一半CNVRs在多数个体中都可检测到。伴随着大量CNVs被发现,鉴定CNV方法的准确性不断被讨论。2008年,Perry等[23]研究了2 191个已被报道的CNV区域,重新检测只确认了其中的1 153个CNVs,并发现这些被确认的CNVs长度中竟然有876个只有报道长度的一半。2015年,Sudmant等[12]对125个不同人类族群共236个个体基因组进行测序,在常染色体和X染色体上分别找到14 467个和545个CNV区域。在黑猩猩[24,25]、果蝇[26]、小鼠[27]、拟南芥[28]、玉米[29]、线虫[30]等模式生物基因组研究中也发现了大量CNVs的存在。
另外,越来越多的报道证实了CNV具有重要功能。在对亚洲人、非洲人和欧洲人的CNV进行通路富集分析研究中发现,22%的通路含有至少一个CNV基因,这些基因大部分属于信号转导通路,6%和表达量有显著相关性[31]。在人类基因组研究中已发现许多疾病与CNVs有关[32]。例如,CCL3L1基因拷贝数低于正常人群的个体更容易感染艾滋病毒[33];FCGR3B基因拷贝数变异与系统性红斑狼疮病显著相关[34];APP基因拷贝数的增加很可能是阿尔兹海默症的致病原因之一[35]。不仅如此,其他一些重要性状也可能是由CNV引起的,在研究其他表型变异及物种进化相关的遗传变异时,我们必须考虑CNV,否则可能会错过重要的遗传信息[36]。
2 CNV在家养动物中的研究
目前已有大量关于畜禽基因组SNP的研究,而CNV的相关研究则显得相对滞后。CNV可作为强有力的分子标记用于畜禽遗传育种。在家养动物牛、羊、鸡、猪中已鉴定出了一些具有代表性的CNVs,更多的CNVs有待进一步挖掘。
2.1 牛
Liu等[14]在11个品种90头牛中鉴定出200个候选拷贝数变异区域(Copy number variation Regions,CNVRs),其中177个区域定位到了染色体上,覆盖了28.1 Mb序列,约占全基因组的1.07%,其中包含400个已注释基因,这些基因与免疫、泌乳、繁殖及反刍等性状相关。随后,2012年该团队在肠道线虫研究中,根据肠道线虫抗性和易感将牛分为两组,每组100个个体,在两组中分别鉴定出297和282个CNVs[37]。Bickhart等[13]用第二代测序技术在5头不同品种的牛中鉴定出1 265个CNVRs。此外,他们还鉴定出一些品种间特异的拷贝数变异,如CATHL4、ULBP17基因拷贝数变异,这可能是不同品种适应性、抗病性、繁殖性能方面存在差异的原因。
牛的繁殖、免疫、泌乳等性状在生产中备受关注,近年来科研工作者在牛基因组发现一些与重要性状相关的CNVs。MIMT1基因中一段110 kb的缺失可导致牛的流产和死胎[38];CLDN16基因中37 kb或56 kb的片段缺失与牛的肾小管发育异常密切相关[39,40];SLC4A2基因编码一种阴离子交换蛋白,该蛋白对破骨细胞维持正常功能是必需的。其外显子2及部分外显子3中约2.8 kb的缺失突变可能导致蛋白功能受到影响,与牛骨硬化病的产生有关[41]。
2.2 羊
2011年,Fontanesi等[15]利用牛的比较基因组芯片用于绵羊拷贝数变异研究,将绵羊基因组与牛的比较基因组芯片进行杂交,共鉴定出135个CNVRs,总长10.5 Mb,占全基因组的0.398%,平均长度为77.6 kb。由于牛羊基因组间的差异,上述研究获得的CNVs较少,不足以构建完整全面的羊CNV图谱。随后,Liu等利用SNP芯片在71只苏尼特羊中鉴定出134个CNVRs,包含800个CNVs,共注释出640个功能基因。对检测到的CNVRs注释基因进行GO富集分析,结果显示这些拷贝数变异区域包含很多与嗅觉、化学刺激感觉等环境应答有关的基因。
ASIP基因[42]具有调控黑色素形成的作用,与动物毛色有关,研究表明绵羊中的ASIP基因发生了190 kb的重复[43]。Fantanesi等[44]对山羊ASIP基因中的拷贝数变异做了进一步研究,他们发现不同品种山羊由于ASIP基因拷贝数的变异和错义突变导致了不同的毛色,包含ASIP基因的片段重复会导致白色毛色。2015年,Dong等[45]在研究中也检测到与绵羊基因组中大小相同的包含有ASIP基因的拷贝数变异与山羊毛色的关联作用。
2.3 猪
2008年,Fadista等[46]首次报道了在猪部分染色体上存在CNV,并在4、7、14和17四条染色体上鉴定出37个CNVRs。之后,Ramayo-Caldas等[47]在伊利比亚猪和长白猪杂交品系中找到49个CNVRs。2012年,Chen等[17]在18个品种的1693头猪中找到了大量CNVs,鉴定出了565 个CNVRs,包含2 122个CNVs。通过QTL分析,筛选出了7个关于猪体尺长、背膘厚度、腹脂重、糖分解能力等表型多态的候选CNVs。
Seo等[48]对位于猪8号染色体的KIT基因研究发现,其全部或局部的基因复制与猪毛色的白色显性有着密切的关系。KIT 是已知的控制毛色表型的基因之一,拷贝数的增加会影响其作为受体的生物活性,造成黑色素细胞前体物不迁移或者部分迁移,导致猪毛不呈色或部分呈色。
2.4 鸡
2010年,Wang等[49]用全基因组Tiling芯片在3个品种的10只鸡中鉴定出96个CNVs,覆盖了16 Mb基因组序列,占全基因组的1.3%。其中多数小片段CNVs是非编码序列,而大片段CNVs多包含基因,他们在此研究基础上构建了第一个鸡的CNV图谱。随后,Wang等[50]通过基因组杂交芯片技术鉴定出了130个CNVRs,包含238个CNVs,使鸡的CNV图谱得到进一步完善。为了获得全面、覆盖率高的拷贝数变异图谱,Crooijmans等[19]对15个品系64只鸡中的CNV进行研究,鉴定出1 556个CNVRs共 3 154个CNVs,覆盖了全基因组的5.4%。上述研究均是基于芯片技术,2014年,Yi等[20]用测序技术完成了12只不同品种的鸡的重测序,找到的CNVRs数量达8 840个,总长98.2 Mb,占基因组的 9.4%。大量的CNVs中极有可能存在对表型差异有显著影响的位点,这有利于研究者们下一步对表型多态相关的基因组水平变异进行筛选。
鸡的豆冠是一个显性突变,该突变使鸡冠和肉垂的体积明显减少,因此豆冠可以有效减少热量的损失并降低鸡冻伤的敏感性,是鸡在寒冷气候下的一种适应性性状。Wright等[51]的研究表明,鸡的豆冠表型是由一段重复序列的大量扩增导致的,该序列靠近编码SOX5转录因子基因的第一内含子,是进化非常保守的非编码序列。该CNV影响了SOX5基因在间充质细胞中的表达,间充质细胞位于外胚层下方,豆冠和肉垂即由此形成。
鸡羽毛的生长相关基因定位于Z染色体上的K基因座位,Bu等[52]和Elferink等[53]都在K基因座中发现了一段串联重复序列,导致PRLR 和SPEF2基因上176 kb的片段重复,慢羽性状与此片段重复密切关联。
3 CNV研究面临的问题
CNV检测是全基因组CNVs研究的起始环节,因此CNV检测方法和策略的完善至关重要。2010年以前,CNV的检测大部分采用CGH和SNP芯片。然而,芯片检测到的CNV数量受限于探针的密度,早期许多研究中发现的CNV一般不过几百个。提高芯片的探针密度后获得的CNV数量在一定程度上得到了提高,但仍然有限。目前更迫切的工作是开辟新的方法检测出更多数量的CNV区域,绘制出更准确、清晰度更高的 CNV 图谱,DNA 测序技术的发展为CNV的检测提供了新的思路和方法。
1977年第一代测序技术诞生,经过近40年的发展,目前测序技术已经发展到了第三代。二代测序技术是现阶段动植物基因组研究中广泛应用的DNA测序技术,如基因组从头构建、重测序、SNP研究、转录组及表达谱分析、转录调控研究。二代测序最显著的特征是高通量,一次能对几十万到几百万条DNA分子进行序列测定,使得对一个物种的转录组测序或基因组深度测序变得方便易行[46]。CNV的研究也多采用二代测序技术,与芯片技术相比测序具有以下优点:(1)提高了CNV的分辨率,可以发现较短的CNV;(2)可以确定CNV的边界;(3)可以检测出个体CNV的绝对拷贝数;(4)对于结构变化复杂的CNV有较高检测效力。
三代测序技术是近几年来发展起来的新技术,其主要特点是单分子测序、准确性更高,在读长上具有极大的优势,对基因组中的复杂结构部分具有较高的辨识度,可以大大提高CNV的检测灵敏性和准确性。然而目前三代测序成本远高于二代,对动物基因组在群体水平进行大规模三代测序鉴定CNV区域,在短时间内较难实现。
测序技术在未来还有极大的发展空间,测序成本也将随之降低,使我们可以用较低成本和更短时间获得更准确的基因序列,对基因组有更深入的了解,更多的SNPs和CNVs等新的变异位点也将被发现。而在大群体中检测少数已知位置的CNV区域是否存在变异,芯片技术目前依然在检测和分析的成本上具有优势,更有利于大规模的育种产业应用。
4 CNV育种应用的分析与展望
4.1 现代育种与分子标记
传统的育种方法为表型育种,然而表型选育周期长、成本高,育种效率不高,难以在短期内取得遗传进展[54]。分子育种不易受环境的影响,且没有性别、年龄的限制,因而允许进行早期选种,克服了传统育种方法周期长、预见性差、准确率低的局限性,可缩短世代间隔,提高选择强度,从而提高选种的效率和准确性[55]。全基因组选择作为分子育种新策略在奶牛育种中已初见成效,常规的乳用种公牛选育主要通过后裔测定,经繁殖获取公牛后代的生产或其他性能进而实现对种公牛遗传价值的评定。该方法周期长、投入大,通常在种公牛出生5年后才能够完成其主要性状的遗传评定。而全基因组选择则是通过检测覆盖全基因组的分子标记,利用基因组水平的遗传信息对个体进行遗传评估,可对青年公牛进行准确预测。全基因组选择实现了乳用种公牛的早期选择,可将选择年龄提前3年,遗传进展速度提高两倍且极大减少了育种成本[56]。
此外,我们还可用直接改变遗传物质(Genetic modification,GM)的方法进行育种。具体方法目前主要有两种,分别是转基因和基因编辑。转基因即通过基因打靶、克隆等技术将已知功能的基因转入特定生物中,与其自身基因组重组,以期获得具有目标性状并可稳定遗传的个体。转基因技术在改良家畜生产性状、提高家畜抗病力等方面显示了广阔的应用前景。基因编辑是近几年来发展起来的可以对基因组完成精确修饰的一种技术,可完成基因定点InDel突变、敲入、多位点同时突变和小片段的删失等,可在基因组水平上进行精确的基因编辑。在基因编辑过程中,没有引入新的基因,安全性有所提高。
得益于动物基因组研究的飞速发展,以DNA多态性为基础的分子遗传标记得到了大量的发掘和应用,分子标记选择育种逐渐发展起来。早期DNA 分子标记包括限制性片段长度多态性(Restriction fragment length polymorphism,RFLP)和基于PCR 技术的分子标记,后者包括随机扩增多态性(Random amplified polymorphic DNA,RAPD)和基于一代测序的简单序列重复(Simple sequence repeat,SSR)标记、InDel 标记、SNP标记等[57]。目前基于芯片和二代测序检测的SNP标记已经成为分子辅助育种中主要的遗传标记。理想的分子标记应具有多态性高、遍布整个基因组、检测手段简单快速、成本低廉、重复性好等特点。近几年发展起来的CNV作为基因组变异的一种重要形式具有多态性高、遍布整个基因组特点,检测手段也在不断多样化。因此,CNV未来可作为分子育种中一种有强有力的分子标记。
而GM动物的安全性一直以来备受争议,但不可否认这种直接改变遗传物质的技术效率更高,我们可以在极短的时间内获得具有预想性状的个体。随着大量相关研究开展和深入,转基因及基因编辑水平也将不断提高,在家养动物遗传育种方面的应用潜力巨大。目前基因编辑技术应用于遗传育种的最主要制约因素是,现有研究无法精确提供大量决定关键经济性状的功能变异,特别是被证明是安全的功能变异。而通过大规模筛查已经存在于家养动物群体中较高频率的SNP和CNV变异,将大大加速基因编辑技术的育种应用范围。
4.2 CNV育种应用展望
近年来,生物技术不断革新,有两大进展尤为突出。第一,基因组研究和生物信息技术发展迅速,我们已经可以快速解析海量基因组或转录组学数据,生物学研究进入了大数据时代。第二,新型的遗传操作和基因组编辑技术层出不穷,克服了经典基因操作中存在的准确性低、效率低等缺点。
随着基因组研究和现代生物技术的发展,分子育种及GM育种在家养动物育种中显示出强大的生命力,逐渐成为动物育种的趋势和主流。分子育种技术、转基因及基因编辑技术是建立在对全基因组具有全面深刻了解的基础之上的,我们必须挖掘存在于全基因组范围的遗传变异。
研究者对于家养动物基因组SNP展开了大量研究,在猪、牛、羊等基因组中均发现了数以百万计的SNP位点,SNP已成为家养动物育种中非常重要的分子标记,且部分已应用于育种中。但数量巨大的SNPs中仅有小部分属于关键的功能变异,毕竟单位点对基因组序列、基因表达调控的影响是有限的。另外,由于连锁关系,在长连锁区域的大量变异中精确定位直接造成某一表型改变的位点并不容易。这些因素均在一定程度上限制了SNPs在育种中的应用,SNPs的研究和应用相对进入了瓶颈阶段。
与此同时,拷贝数变异研究逐渐成为了动植物基因组研究的热点。CNV普遍存在于动植物基因组中,在家养动物中也发现了大量的CNVs,并鉴定出了一些与重要性状相关的变异。与SNPs相比,CNVs在数量上相对较少,但这从定位上来说无疑是一种优势。而且,CNVs片段长度大,覆盖了更大范围的基因组序列,与SNPs相比其影响基因表达并造成表型差异的概率更高。因此,CNV可以作为一种强有力的分子标记应用于畜禽的选种育种。在育种过程中,对于与某项表型性状或者疾病相关的CNV进行分型,在全基因组水平上选取适应性强、功能性好、抗病能力强的群体,为疾病的研究和良好性状选育提供新的思路。另外,基因编辑技术对功能变异位点信息的要求较高,精确定位的CNV也可作为基因编辑的位点。由于基因之间存在强烈的连锁不平衡性,各SNP位点并不是独立存在的,某一位点的改变并不能导致性状差异。相较于此,CNV显示出了一定的优势。我们相信,未来CNV将能更广泛地应用于家养动物的育种工作。
[1] International Chicken Genome Sequencing, Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution[J]. Nature, 2004, 432(7018):695-716.
[2] Consortium BH. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds[J]. Science, 2009, 324(5926):528-532.
[3] Groenen MA, Archibald AL, Uenishi H, et al. Analyses of pig genomes provide insight into porcine demography and evolution[J]. Nature, 2012, 491(7424):393-398.
[4] Dong Y, Xie M, Jiang Y, et al. Sequencing and automated wholegenome optical mapping of the genome of a domestic goat(Capra hircus)[J]. Nature Biotechnology, 2013, 31(2):135-141.
[5] Huang Y, Li Y, Burt David W, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species[J]. Nature Genetics, 2013, 45(7):776-783.
[6] Jiang Y, Xie M, Chen W, et al. The sheep genome illuminates biology of the rumen and lipid metabolism[J]. Science, 2014, 344(6188):1168-1173.
[7] Wang Y, Lu Y, Zhang Y, et al. The draft genome of the grass carp(Ctenopharyngodon idellus)provides insights into its evolution and vegetarian adaptation[J]. Nature Genetics, 2015, 47(6):625-631.
[8] MacDonald JR, Ziman R, Yuen R, et al. The Database of Genomic Variants:a curated collection of structural variation in the human genome[J]. Nucleic Acids Research, 2014, 42(D1):D986-D992.
[9] Rubin CJ, Zody M, Eriksson J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication[J]. Nature, 2010, 464(7288):587-591.
[10] Daetwyler HD, Capitan A, Pausch H, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle[J]. Nature Genetics, 2014, 46(8):858-865.
[11] Carneiro M, Rubin CJ, Di Palma F, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication[J]. Science, 2014, 345(6200):1074-1079.
[12] Sudmant PH, Mallick S, Nelson BJ, et al. Global diversity,population stratification, and selection of human copy-number variation[J]. Science, 2015, 349(6253):aab3761.
[13] Bickhart DM, Hou Y, Schroeder SG, et al. Copy number variation of individual cattle genomes using next-generation sequencing[J]. Genome Research, 2012, 22(4):778-790.
[14] Liu GE, Hou Y, Zhu B, et al. Analysis of copy number variations among diverse cattle breeds[J]. Genome Research, 2010, 20(5):693-703.
[15] Fontanesi L, Beretti F, Martelli PL, et al. A first comparative map of copy number variations in the sheep genome[J]. Genomics,2011, 97(3):158-165.
[16] Ma Y, Zhang Q, Lu Z, et al. Analysis of copy number variations by SNP50 BeadChip array in Chinese sheep[J]. Genomics, 2015,106(5):295-300.
[17] Chen C, Qiao R, Wei R, et al. A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complextraits[J]. BMC Genomics, 2012, 13(1):733.
[18] Li Y, Mei S, Zhang X, et al. Identification of genome-wide copy number variations among diverse pig breeds by array CGH[J]. BMC Genomics, 2012, 13(1):725.
[19] Crooijmans RP, Fife MS, Fitzgerald TW, et al. Large scale variation in DNA copy number in chicken breeds[J]. BMC Genomics,2013, 14(1):398.
[20] Yi G, Qu L, Liu J, et al. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing[J]. BMC Genomics, 2014, 15(1):962.
[21] Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome[J]. Science, 2004, 305(5683):525-528.
[22] Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome[J]. Nature, 2006, 444(7118):444-454.
[23] Perry GH, Ben-Dor A, Tsalenko A, et al. The fine-scale and complex architecture of human copy-number variation[J]. The American Journal of Human Genetics, 2008, 82(3):685-695.
[24] Perry GH, Yang F, Marques-Bonet T, et al. Copy number variation and evolution in humans and chimpanzees[J]. Genome Research, 2008, 18(11):1698-1710.
[25] Perry GH, Ben-Dor A, Tsalenko A, et al. Hotspots for copy number variation in chimpanzees and humans[J]. Proceedings of the National Academy of Sciences, 2006, 103(21):8006-8011.
[26] Meisel RP, Han MV, Hahn MW. A complex suite of forces drives gene traffic from Drosophila X chromosomes[J]. Genome Biology and Evolution, 2009, 1:176-188.
[27] Orozco LD, Cokus SJ, Ghazalpour A, et al. Copy number variation influences gene expression and metabolic traits in mice[J]. Human Molecular Genetics, 2009, 18(21):4118-4129.
[28] Ossowski S, Schneeberger K, Clark RM, et al. Sequencing of natural strains of Arabidopsis thaliana with short reads[J]. Genome Research, 2008, 18(12):2024-2033.
[29] Springer NM, Ying K, Fu Y, et al. Maize inbreds exhibit high levels of copy number variation(CNV)and presence/absence variation(PAV)in genome content[J]. PLoS Genet, 2009, 5(11):e1000734.
[30] Maydan JS, Lorch A, Edgley ML, et al. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans[J]. BMC Genomics, 2010, 11(1):62.
[31] Poptsova M, Banerjee S, Gokcumen O, et al. Impact of constitutional copy number variants on biological pathway evolution[J]. BMC Evolutionary Biology, 2013, 13(1):19.
[32] Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome[J]. Nature, 2010,464(7289):704-712.
[33] Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility[J]. Science, 2005, 307(5714):1434-1440.
[34] Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans[J]. Nature, 2006, 439(7078):851-855.
[35] Theuns J, Brouwers N, Engelborghs S, et al. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease[J]. The American Journal of Human Genetics, 2006, 78(6):936-946.
[36] Komura D, Shen F, Ishikawa S, et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays[J]. Genome Research, 2006, 16(12):1575-1584.
[37] Hou Y, Liu GE, Bickhart DM, et al. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle[J]. Functional & Integrative Genomics, 2012, 12(1):81-92.
[38] Flisikowski K, Venhoranta H, Nowacka-Woszuk J, et al. A novel mutation in the maternally imprinted PEG3 domain results in a loss of MIMT1 expression and causes abortions and stillbirths in cattle(Bos taurus)[J]. PLoS One, 2010, 5(11):e15116.
[39] Müller D, Kausalya PJ, Claverie-Martin F, et al. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting[J]. The American Journal of Human Genetics, 2003, 73(6):1293-1301.
[40] Testoni S, Mazzariol S, Drögemüller C, et al. Renal dysplasia in grey Alpine breed cattle unrelated to CLDN16 mutations[J]. Veterinary Record, 2012, 170(1):22.
[41] Meyers SN, McDaneld TG, Swist SL, et al. A deletion mutation in bovine SLC4A2 is associated with osteopetrosis in Red Angus cattle[J]. BMC Genomics, 2010, 11(1):337.
[42] Bultman SJ, Michaud EJ, Woychik RP. Molecular characterizationof the mouse agouti locus[J]. Cell, 1992, 71(7):1195-1204.
[43] Norris BJ, Whan VA. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep[J]. Genome Research, 2008, 18(8):1282-1293.
[44] Fontanesi L, Beretti F, Riggio V, et al. Copy number variation and missense mutations of the agouti signaling protein(ASIP)gene in goat breeds with different coat colors[J]. Cytogenetic and Genome Research, 2009, 126(4):333-347.
[45] Dong Y, Zhang X, Xie M, et al. Reference genome of wild goat(Capra aegagrus)and sequencing of goat breeds provide insight into genic basis of goat domestication[J]. BMC Genomics, 2015,16(1):431.
[46] Fadista J, Nygaard M, Holm LE, et al. A snapshot of CNVs in the pig genome[J]. PLoS One, 2008, 3(12):e3916.
[47] Ramayo-Caldas Y, Castelló A, Pena RN, et al. Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip[J]. BMC Genomics, 2010, 11(1):593.
[48] Seo BY, Park EW, Ahn SJ, et al. An accurate method for quantifying and analyzing copy number variation in porcine KIT by an oligonucleotide ligation assay[J]. BMC Genetics, 2007, 8(1):81.
[49] Wang X, Nahashon S, Feaster TK, et al. An initial map of chromosomal segmental copy number variations in the chicken[J]. BMC Genomics, 2010, 11(1):351.
[50] Wang Y, Gu X, Feng C, et al. A genome-wide survey of copy number variation regions in various chicken breeds by array comparative genomic hybridization method[J]. Animal Genetics,2012, 43(3):282-289.
[51] Wright D, Boije H, Meaclows JR, et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens[J]. PLoS Genet, 2009, 5(6):e1000512.
[52] Bu G, Huang G, Fu H, et al. Characterization of the novel duplicated PRLR gene at the late-feathering K locus in Lohmann chickens[J]. Journal of Molecular Endocrinology, 2013, 51(2):261-276.
[53] Elferink MG, Vallée A, Jungerius AP, et al. Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken[J]. BMC Genomics, 2008, 9(1):391.
[54] Stuber CW, Polacco M, Senior ML. Synergy of empirical breeding,marker-assisted selection, and genomics to increase crop yield potential[J]. Crop Science, 1999, 39(6):1571-1583.
[55] Moose SP, Mumm RH. Molecular plant breeding as the foundation for 21st century crop improvement[J]. Plant Physiology, 2008,147(3):969-977.
[56] Schaeffer L. Strategy for applying genome-wide selection in dairy cattle[J]. Journal of Animal Breeding and Genetics, 2006, 123(4):218-223.
[57] 黎裕, 贾继增, 王天宇. 分子标记的种类及其发展[J]. 生物技术通报, 1999(4):19-22.
(责任编辑李楠)
Progress and Prospects in Domestic Animals and Breeding:a Review of Genomic Copy Number Variations
Cheng Hong Jiang Yu
(College of Animal Science and Technology,Northwest A&F University,Yangling 712100)
The significant progress of de novo assembly and re-sequencing of agricultural animal genomes, promoted the discovery of abundant genetic variations. Global variations mainly include single nucleotide polymorphisms(SNPs)and copy number variations(CNVs). Unlike SNPs, which have been widely studied and applied as molecular marker in breeding, fewer CNVs have been detected and experimental validated. Therefore, CNVs rarely serve as molecular markers in breeding. However, the proportion of genome variation ascribed to CNVs are far larger than to SNPs. So CNVs may have great impacts on agronomic traits. Recently, CNV is on the cutting edge of the research studies as well as applications in livestock and poultry breeding. In this review, we discuss the research progress of CNV, and its prospects for breeding domestic animals.
domestic animal;genome;copy number variation;breeding;economic trait
10.13560/j.cnki.biotech.bull.1985.2015.11.006
2015-09-27
陕西省科技新星资助项目(K3320215201)
程红,女,硕士研究生,研究方向:动物遗传;E-mail:chenghong92@163.com
姜雨,男,教授,博士生导师,研究方向:动物遗传;E-mail:jiangyu96@163.com
猜你喜欢
河北医学(2021年10期)2021-10-27
中国临床医学影像杂志(2019年6期)2019-08-27
百科知识(2018年9期)2018-05-28
现代园艺(2017年21期)2018-01-03
小学生作文·小学低年级适用(2016年8期)2017-01-16
中华实验和临床病毒学杂志(2016年3期)2016-08-09
中国康复理论与实践(2015年10期)2015-12-24
读写算·高年级(2015年4期)2015-07-25
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年5期)2015-02-06
