
人类长链非编码RNA相关SNP鉴定与功能预测的研究进展
2015-09-14 12:47:12龚静柳纯洁缪小平郭安源
生物技术通报 2015年11期
龚静 柳纯洁 缪小平 郭安源
随着高通量测序技术的发展,人们发现人类基因组中绝大部分DNA可转录为RNA,但其中能够编码蛋白质(Protein-coding)的DNA仅占全基因组很少一部分(约2%)[1],剩余绝大部分DNA也可以转录为不能翻译成蛋白质的RNA,即非编码RNA(Non-coding RNA,ncRNA)[2]。 根 据 RNA 长度,非编码RNA又主要分为短链RNA(small RNA,smRNA)和长链非编码RNA(Long non-coding RNA,lncRNA)。smRNA主要包括转录起始RNA(tiRNA,18 nt)、Piwi蛋 白 相 互 作 用 RNA(piRNA,26-31 nt)、微小 RNA(microRNA,22 nt)、小核仁 RNA(snoRNA,60-300 nt)等。lncRNA是一类长度大于200个核苷酸,不表现蛋白质编码潜能的RNA[3]。lncRNA作为一种ncRNA,一直被认为是基因转录“噪音”而未受重视。然而,最近的研究表明,lncRNA在正常发育和疾病发生发展过程中都扮演着重要的角色,具有丰富的生物学功能:参与X染色体的失活[4],调控mRNA的降解[5],参与造血系统及免疫应答[6,7]、构成细胞核亚结构的结构骨架[8],作为染色质重塑(Chromatin remodeling)调控因子[9,10]等。
单核苷酸多态性(Single nucleotide polymorphism,SNP)是指在基因组DNA水平上发生的单个核苷酸变异所引起的DNA序列多态性[11,12]。随着人类基因组计划和千人基因组(1000 genome)项目的完成[12,13],人类基因组基本趋于完善。目前已知人类基因组大约有30亿对碱基,其中可能发生变异的位点多达88 111767(NCBI dbSNP 142),平均每60个碱基就可能出现一个SNP。研究表明健康个体之间基因组碱基差异大约为0.1%,即两个随机个体中每1200-1500个碱基就会有一个差异碱基。这些SNPs不仅是造成健康个体间差异的重要因素,而且大量研究还证实,有些SNP与疾病易感性、药物敏感性及疾病发生发展都有关系[14,15]。随着SNP分型技术的发展,SNP标记的发现和定位越来越多。相对于以限制性片段长度多态性为代表的第一代遗传标记和微卫星多态性为代表的第二代遗传标记,SNP具有分布广泛、数量多等特点,成为了第三代遗传标记,更加适合于基因性状及疾病的研究[16]。
既然lncRNA不是垃圾基因,那么lncRNA上的SNP也有可能通过改变lncRNA的功能而成为功能性SNP。基于这个假设,很多研究者展开了一系列工作并取得了一定的成果。本文拟对这些lncRNA相关SNP文献进行综述,介绍lncRNA相关SNP的鉴定与功能预测方法以及相关生物信息数据库,供相关研究者参考,以期能够为研究lncRNA提供新的策略。
1 SNP常用研究方法
目前国内外SNP研究方法大致有如下三种。
1.1 分子流行病学研究
可分为全基因组关联分析(Genome-wide associ-ation study,GWAS)和候选基因策略。其中,GWAS是以全基因组SNP标记为研究内容进行病例-对照关联分析,以期发现影响疾病或者复杂性状遗传特征的一种策略,该方法在研究性状相关SNP方面已取得了重大成果。美国国家人类基因组研究所(NHGRI)对所有发表的GWAS文献进行整理,建立了一个在线实时更新网站(NHGRI GWAS Catalog,https://www.genome.gov/26525384, 现 已 移到EBI http://www.ebi.ac.uk/gwas/),可以按照疾病方便地下载GWAS鉴定的SNP[17]。截止2015年2月,该数据库已收录了15 000多个与各种性状相关的SNP,这些性状既包括单基因疾病,也包括癌症、肥胖症、糖尿病、精神分裂症、高血压、老年痴呆症等复杂疾病[18-21]。这些SNP有助于确定基因多态性与疾病的关系,解释个体间表型差异对疾病易感程度,研究不同基因型个体对药物反应差异和指导药物开发及临床合理用药等。然而,GWAS也存在一定的局限性。虽然GWAS成本有所下降,但仍需耗费大量精力和经费。另外,GWAS也并非对所有SNP进行研究,而是先根据单体型图谱和连锁不平衡关系在全基因组范围内选择标签SNP(tagSNP),其实验所发现的疾病相关SNP只能代表其连锁的区域与疾病有关系,而真正的“致病”遗传变异(Causal genetic variants)还有待进一步精细分析。因此,仍有很多研究者采用候选基因和候选通路策略进行分子流行病学研究。在GWAS研究还未兴起时,传统的筛选策略发挥了重要作用,发现了大量疾病相关SNP。在后GWAS时代,很多研究者把GWAS发现的易感区域作为候选位点,然后对该区域进行精细定位或者功能实验,并获得了重要的研究成果。
1.2 SNP相关的生物信息学研究
生物信息学(Bioinformatics)作为一门交叉学科,在SNP的鉴定、注释、储存、功能预测等各个方面发挥重要功能。如 SAMtools[22]、GATK[23]工具可以从全基因组DNA测序和外显子DNA测序中鉴定SNP ;ANNOVAR[24]、SIFT[25]、SNPinfo[26]工具 可以对SNP进行注释,寻找其潜在的功能;dbSNP用于SNP的储存;plink[27]工具用于SNP与疾病相关性的分析,miRNASNP[28]、PolymiRTS[29]数据库可以方便地搜索miRNA相关的数据库等。
1.3 SNP功能实验
无论是人群研究得到的疾病相关位点还是生物信息学预测的功能性SNP,最终都需要通过分子生物学实验探索其具体的生物作用机制。最常用的方法是构建野生型和变异型载体,将其转染于细胞,通过荧光素酶报告基因实验验证SNP对基因表达水平的影响,运用RT-PCR,Western blot等方法检测野生型和变异型细胞中靶基因mRNA和蛋白质表达水平。同时还可以观察转染后细胞的生物学特性的变化:细胞生长与增殖、细胞凋亡与细胞周期分布、细胞迁移能力等。
2 分子流行病学发现的lncRNA相关SNP
H19基因是最早被发现的有功能的lncRNA之一,位于人染色体11p15.5,编码一个2.3 kb的lncRNA。Petry等[30]选取了H19基因上的3个SNP位点,对一个出生队列中的1696名儿童、822名母亲和661名父亲进行基因分型。结果显示,孩子和母亲的H19 2992 C>T SNP基因型与子代出生体重(P=0.03)相关。母亲的基因型也与脐带血IGF-II 水平相关(P=0.0003)。Verhaegh 等[31]通过Haploview软件在H19基因和上游启动子区选择了5个tagSNP,通过病例对照研究,并使用逻辑回归分析来评估这些SNPs与癌症风险的关联,最终发现rs2839698 TC(OR=0.60,95% CI=0.36-0.99)基 因型可以明显减少膀胱癌的发病风险。
HOX转录反义RNA(HOX transcript antisense RNA,HOTAIR)是另一个研究较多的lncRNA。HOTAIR定位于HOXC基因座12q13.13。HOTAIR的5'端可招募结合多梳蛋白抑制复合物2(polycomb repressive complex 2,PRC2), 借 助 PRC2上 3个H3K27 甲 基 转 移 酶 EZH2、SUZ12 和 EED[9], 使另一基因座HOXD上长约40 kb序列转录沉默,从而使乳腺上皮细胞倾向于胚胎成纤维细胞样表型。HOTAIR上SNP在不同样本中的病例-对照研究发现,HOTAIR基因上的rs920778与乳腺癌[32]、食管癌[33]、胃癌[34]的发病风险都相关。在中国济南和淮安人群中,rs920778 TT携带者是CC携带者患胃癌风险的1.66倍和1.87倍。在中国济南、石家庄、淮安地区的人群中,rs920778 TT携带者比CC携带者患食管癌的风险高1.37倍、1.78倍和2.08倍。
3 生物信息学在lncRNA相关SNP中的应用
lncRNA SNP的确在疾病的发生发展中扮演重要角色,那么如何全面地挖掘lncRNA相关SNP,以及如何在众多lncRNA相关的SNP中筛选一定数量SNP进行功能实验还需要借助生物信息学方法。下面就将生物信息学在lncRNA相关SNP中的应用展开详细综述。
3.1 lncRNA数据资源
lncRNA数量越多,相关SNP数量也越多,选择不同的lncRNA数据库也会影响SNP的鉴定数目。因此,我们先对目前可用的lncRNA相关数据库进行综述。早期发现的lncRNA分散在NCBI GenBank、UCSC、Ensembl这些大型数据库中,RNAdb是最早出现的系统性收集ncRNA的数据库,该数据库包含了800多条实验验证的ncRNAs以及从人和老鼠的cDNA 中预测了 20 000多条 ncRNA[35]。lncRNAdb是最早从文献中人工收集真核生物中lncRNA信息的数据库[36],而GENCODE相对来说是使用较多的一个含有lncRNA数据的网站,最新的版本(v22)已含有15 900条lncRNA基因,27 670条长非编码转录本[37]。另外一个常用的资源,LNCipedia含有32108条注释的人类lncRNA转录本[38],这个数据库预测了每个转录本的二级结构及编码蛋白质的可能性;基于蛋白组学实验,该数据库还开发了一个流程用于预测lncRNA开放阅读框。NONCODE是中国科学院生物物理研究所和计算所开发的一个ncRNA在线资源,现在已更新到V4版本[39,40]。它不仅提供各个转录本的序列信息,还提供了lncRNA在每个组织的表达量信息。目前NONCODE V4版本中,人类lncRNA基因数量已有54 073条,转录本数量有92343条,小鼠的lncRNA数量已有46 475条,转录本数量有67 628条。lncRNAMap数据库广泛收集了各个公共资源的RNA-seq数据,然后用自主开发的流程进行lncRNA注释及表达量的计算[41]。该网站提供了不同组织、细胞系和疾病状态下的lncRNA表达信息,以及miRNA-lncRNA相互作用关系。另外两个数据库lncRNome[42]和Functional lncRNA[43]数据库通过整合其他数据库资源预测lncRNA功能。lncRNome含有18 000个人类的lncRNA转录本,提供的注释信息包括基因序列、转录本序列、RNA加工信息、miRNA结合位点和lncRNA启动子区域的表观修饰信息。该数据库也把遗传变异的信息加入其中,构建了一个基因组浏览页面。Functional lncRNA数据库包含3个子数据库,人工收集了人、小鼠、大鼠的lncRNA信息。除了直接提供lncRNA序列信息的数据库外,许多其它功能的lncRNA数据库也被随之开发出来,例如提供表达谱信息、RNA相互作用信息或者提供相关疾病信息。Starbase v2.0[44](lncRNABase)通过整合大量高通量测序数据挖掘RNA/RNA、RNA/蛋白质相互作用位点,提供了超过10 000条miRNA-lncRNA相互作用信息。NED网站包含了lncRNA的基因芯片、原位杂交表达量信息、进化保守性及二级结构等信息[45]。
3.2 lncRNA上SNP鉴定
通过比较SNP和lncRNA基因在基因组上的位置,可确定lncRNA上的SNP。lncRNASNP数据库[46](http ://bioinfo.life.hust.edu.cn/lncRNASNP/)是本课题组构建的一个lncRNA相关SNP及其可能功能影响的数据库,其lncRNA数据来源于LNCipedia数据库,包括17 436条人类lncRNA基因的32108条转录本(基因组版本:GRCh37/hg19)。SNP信息来自NCBI dbSNP数据库v138版本。lncRNASNP数据库在lncRNA的外显子区域一共发现了495 729个SNPs。
3.3 lncRNA上SNP功能预测
3.3.1 SNP对lncRNA二级结构的影响 DNA中SNP的存在导致改变RNA的序列,从而影响RNA的二级结构和高级结构。部分lncRNAs在生物学过程中扮演支架的角色[3],因此形成正确的空间结构是lncRNAs发挥功能的基础。有研究者推断lncRNA上的SNP可能影响lncRNA二级结构及其稳定性,从而影响lncRNA的表达和功能[47]。常用的RNA二 级 结 构 预 测 软 件 有 RNAfold[48]、RNAsoft[49]、Mfold[50]等。lncRNASNP数据库使用 RNAfold预测lncRNA外显子上所有SNP对lncRNA二级结构的影响。对于每个SNP,把SNP相应位置的碱基由参考基因型转为另一等位基因型,得到突变型转录本。使用RNAfold对野生型和突变型转录本进行二级结构预测,得到预测的二级结构图和最小自由能(MFE,ΔG)。ΔΔG=|ΔG 突变 -ΔG 野生 |,即为每个 SNP 造成的能量改变。分析的结果显示,SNP造成的平均能量变化为(1.30±1.62)kcal/mol,前10%的能量变化是3.10 kcal/mol。
3.3.2 SNP对miRNA lncRNA相互作用的影响 大量实验证据都表明,miRNA也可以在转录后水平调控lncRNA的表达或者lncRNA通过与miRNA结合影响miRNA对靶基因的调控。Starbase数据库提供了超过10 000条miRNA-lncRNA相互作用信息。miRNA能够调控RNA主要取决于miRNA 5'端前8个碱基与靶基因结合的自由能[51]。如果miRNA与靶基因结合位点上的SNPs能够引起自由能的显著改变或其二级结构的改变,将会影响miRNA与靶序列的有效结合。对于编码基因,已有多个数据库系统地预测了编码基因3'UTR上影响miRNA与靶基因结合的 SNP,如 miRNASNP[28]、MicroSNiPer[52]、RNASNP[53]、PolymiRTS[29]、miRdSNP[54]、MirSNP[55],而对于非编码基因,系统性的数据库还比较少。系统研究影响miRNA与靶基因结合SNP的方法大致为:(1)选取要研究的基因并获得其基因组位置(编码基因或非编码基因),然后把SNP数据比对到这些基因上。(2)对基因上的SNP,截取SNP上下游的基因序列,根据SNP的等位基因型,得到野生型转录本和变异型转录本。(3)利用miRanda[56]、Diana-MicroT[57]、PicTar[58]、TargetScan[56]、MicroInspector[59]等软件预测野生型转录本和变异型转录本上可能存在的miRNA结合位点。(4)比较野生型转录本和变异型转录本miRNA结合情况,得到可能影响miRNA与靶基因结合的SNP。lncRNASNP用miRanda和TargetScan两种预测方法分别对lncRNA上的SNP及其上下游25bp的序列进行miRNA靶位点的分析,预测了大量可能影响miRNA-lncRNA结合的SNP。多数SNP既能造成某些miRNA与lncRNA的结合丧失,同时又能获得一些新的miRNA结合位点。lncRNASNP一共预测262154个SNP可能导致miRNA与lncRNA的结合丧失,280 012个SNP可能获得新的miRNA与lncRNA结合。由于预测的功能性SNP众多,该数据库还对SNP设置了一系列筛选条件。用户可以根据SNP所在序列的保守性,miRNA的表达量,影响的miRNA lncRNA结合实验支持与否进行筛选。目前人类注释的miRNA已有2000多条,但是在单个样本中,每个miRNA的表达量高低不等,约21%的miRNA表达量占总量的90%。剩下的79%表达量很小或者只在特别的组织或者细胞状态下才表达[60]。在研究特定组织时,我们可以只选择该组织相对高表达的miRNA靶位点上的SNP。对于低表达或者不表达的miRNA,即使存在预测的功能性SNP,在实际条件中,发挥的功能也非常有限。
3.4 GWAS与lncRNA SNP
如前文介绍的美国国家人类基因组研究所(NHGRI)对所有发表的GWAS文献进行整理,收集了所有基因分型分析P<1×10-5的SNP,并对这些SNP进行了简单分类,分析(截止到2015年4月7日)发现,大部分GWAS相关SNP都位于基因间区和内含子区,只有很小一部分位于已知基因的编码区(图1)。如何解析这些非编码区的SNP功能是后GWAS研究的一个难点。随着被发现的lncRNA日益增加及其功能注释的越来越多,研究者逐渐把目光投到lncRNA上。早在2011年,Jin等[61]就在GWAS Catalog网站整理的1998个疾病易感区域中发现52个易感位点是在lncRNA区域,风险位点在lncRNA上的富集程度(lncRNA上的风险位点/lncRNA的总长度)是整个基因组(所有风险位点/基因组总长度)的1.5倍,并且发现这种富集在前列腺癌中更为明显。当时GWAS Catalog收集了33个前列腺癌的独立风险位点,其中有8个在lncRNA基因上。Jin等[61]接下来在两个前列腺癌GWAS研究中重新观察lncRNA相关的SNP与前列腺癌的易感相关性,发现93个lncRNA上的SNP与前列腺癌的患病风险相关(P<0.001)。其中有60个落在以前报告的区域,另外33个SNP分布在10个LD区域(Linkage disequilibrium region)。对10个LD区域中各选择一个SNP进行人群验证,发现rs3787016与前列腺癌的发病风险显著相关,合并人群分析的P值达到7.22E-7。
lncRNASNP利用GWAS Catalog数据(截止2014年5月,13 383个疾病相关tagSNP),发现142个GWAS鉴定的tagSNP是落在lncRNA基因上,这些SNP涉及到多种疾病。同时也对tagSNP在不同人种中的LD区域的SNP进行分析,发现有更多的SNP落在lncRNA区域。
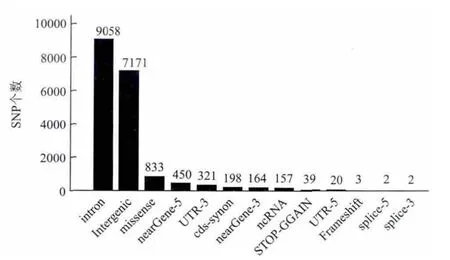
图1 GWAS鉴定的SNP在基因组上的区域分布
3.5 疾病与lincRNA SNP
根据在基因组上与蛋白编码基因的相对位置关系,lncRNA又可分为不同的5个亚类:正义长非编码RNA、反义长非编码RNA、双向的长非编码RNA、内含子长非编码RNA和基因间长链非编码RNA。其中基因间长链非编码RNA(Long intergenic non-coding RNA,lincRNA)是研究得较多的一类lncRNA。2014年,Li等[62]从6个数据库收集了人类128 407个疾病(表型)相关的SNP,然后把这些SNP比对到5 700条人类lincRNA,发现11631个SNP可以比对到3 323条人类的lincRNA上或者其上下游10 kb区域。进一步把疾病相关SNP所在的LD区域中的所有SNP也纳入分析,他们发现了128 785个在lincRNAs附近。他们的研究表明约1/3的lincRNA附近含有疾病相关的SNP,有些lincRNA甚至包含多达6个疾病相关SNP。
4 展望
目前,对于lncRNA相关SNP的研究还处于起始阶段。虽然有一些数据库对lncRNA上的SNP进行了系统性的挖掘,但对这些SNP的潜在功能分析还非常有限。除了SNP对lncRNA基因二级结构和miRNA lncRNA结合的影响外,lncRNA上还可能存在其他功能性SNP,如lncRNA上游也存在许多基因修饰位点和转录因子结合位点,这些区域的SNP也有可能影响lncRNA的基因转录。另外,lncRNA的功能中包括与miRNA相互结合和与其它DNA/RNA结合,因此,理论上还会存在很多非lncRNA上的SNP可能影响lncRNA的功能。例如,在miRNA相关SNP的研究中,学者不仅关注miRNA基因上的SNP,miRNA靶基因上的SNP常常也是研究的重点。最后,lncRNA上SNP功能的实现主要还与lncRNA本身的功能有关,而lncRNA又具有类型多、作用模式多和数量多的特点,目前对lncRNA进行完善注释的非常少。因此,研究lncRNA及相关SNP在疾病中的作用机制,是未来的重要研究方向之一。
[1]Qi P, Du X. The long non-coding RNAs, a new cancer diagnostic and therapeutic goldmine[J]. Modern Pathology:an Official Journal of theUnited States and Canadian Academy of Pathology, Inc, 2013,26(2):155-165.
[2]Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project[J]. Nature, 2007, 447(7146):799-816.[3]Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs:insights into functions[J]. Nature Genetics, 2009, 10(3):155-159.[4]Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA[J].Nature Genetics, 2002, 30(2):167-174.
[5]Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3'UTRs via Alu elements[J]. Nature,2011, 470(7333):284-288.
[6]Carpenter S, Aiello D, Atianand MK, et al. A long noncoding RNA mediates both activation and repression of immune response genes[J]. Science, 2013, 341(6147):789-792.
[7]Han BW, Chen YQ. Potential pathological and functional links between long noncoding RN As and hematopoiesis[J]. Science Signaling, 2013, 6(289):re5.
[8]Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA:NEAT1 RNA is essential for the structure of paraspeckles[J]. Molecular Cell, 2009, 33(6):717-726.
[9]Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis[J].Nature, 2010, 464(7291):1071-1076.
[10]Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes[J]. Science, 2010,329(5992):689-693.
[11]Carlson CS, Eberle MA, Kruglyak L, et al. Mapping complex disease loci in whole-genome association studies[J]. Nature,2004, 429(6990):446-4 52.
[12]Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing[J]. Nature, 2010,467(7319):1061-10 73.
[13]Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1, 092human genomes[J]. Nature, 2012,491(7422):56-65.
[14]Hovhannisyan Z, We iss A, Martin A, et al. The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease[J]. Nature, 2008, 456(7221):534-53 8.
[15]Hruska KS, LaMarca ME, Scott CR, et al. Gaucher disease:mutation and polymorphism spectrum in the glucocerebrosidase gene(GBA)[J]. Human Mutation, 2008, 29(5):567-583.
[16]Garvin MR, Sa itoh K, Gharrett AJ. Application of single nucleotide polymorphisms to non-model species:a technical review[J].Molecular Ecology Resources, 2010, 10(6):915-93 4.
[17]Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations[J]. Nucleic Acids Research, 2014, 42(Database issue):D1001-1006.
[18]Todesco M, Balasubramanian S, Hu TT, et al. Natural allelic variation underlying a major fitness trade-off in A rabidopsis thaliana[J]. Nature, 2010, 465(7298):632-636.
[19]Bochukova EG, Huang N, Keogh J, et al. Large, rare chromosomal deletions associated with severe early-onset obesity[J]. Nature,2010, 463(7281):666-670.
[20]Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity[J]. Nature, 2010, 466(7302):113-117.
[21]Lango Allen H, Estrada K, Lettre G, et al. Hundreds of variants clustered i n genomic loci and biological pathways affect human height[J]. Nature, 2010, 467(7317):832-838.
[22]Ramirez-Gonzalez RH, Bonnal R, Caccamo M, et al. Bio-samtools:Ruby bindin gs for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments[J]. Source Code for Biology and Medicine, 2012, 7(1):6.
[23]McKenna A, Hanna M, Ba nks E, et al. The Genome Analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data[J]. Genome Research, 2010, 20(9):1297-1303.
[24]Wang K, Li M, H akonarson H. ANNOVAR :functional annotation of genetic variants from high-throughput sequencing data[J].Nucleic Acids Research, 2010, 38(16):e164.
[25]Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm[J]. Nature Protocols, 2009, 4(7):1073-1081.
[26]Xu Z, Taylor JA. SNPinfo:integrating GWAS and candidate gene information into functional SNP selection for genetic association studies[J]. Nucleic Acids Research, 2009, 37(Web S erver issue):W600-605.
[27]Purcell S, Neale B, Todd-Brown K, et al. PLINK :a tool set for whole-genome association and population-based linkage analyses[J]. American Journal of Human Genetics, 2007, 81(3):559-575.
[28]Gong J, Tong Y, Zhang HM, et al. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis[J]. Human Mutation, 2012, 33(1):254-263.
[29]Bhattacharya A, Ziebarth JD, Cui Y. PolymiRTS Database 3. 0:linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways[J]. Nucleic Acids Res earch, 2014, 42(Database issue):D86-91.
[30]Petry CJ, Ong KK, Barratt BJ, et al. Common polymorphism in H19 associated with birthweight and cord blood IGF-II levels in humans[J]. BMC Genetics, 2005, 6:22.
[31]Verhaegh GW, Verkleij L, Vermeulen SH, et al. Polymorphisms in the H19 gene and the risk of bladder cancer[J]. EuropeanUrology, 2008, 5 4(5):1118-1126.
[32]Bayram S, Sumbul AT, Batmaci CY, et al. Effect of HOTAIR rs920778 polymorphism on breast cancer susceptibility and clinicopathologic features in a Turkish population[J].Tumour Biology:the journal of the International Society for Oncodevelopmental Biology and Medicine, 2015, 36(5):DOI: 10. 1007/s13277-014-3028-0.
[33]Zhang X, Zhou L, Fu G, et al. The identification of an E SCC susceptibility SNP rs920778 that regulates the expression of lncRNA HOTAIR via a novel intronic enhancer[J].Carcinogenesis, 2014, 35(9):2062-2067.
[34]Pan W, Liu L, Wei J, et al. A functio nal lncRNA HOTAIR genetic variant contributes to gastric cancer susceptibility[J]. Molecular Carcinogenesis, 2015:DOI: 10. 1002/mc. 22261.
[35]Pang KC, Stephen S, Engstrom PG, et al. RNAdb--a comprehensive mammalian noncoding RNA database[J]. Nucleic Acids Research, 2005, 33(Database issue):D125-130.
[36]Quek XC, Thomson DW, Maag JL, et al. lncRNAdb v2. 0:expanding the reference database for functional long noncoding RNAs[J]. Nucleic Acids Research, 2015, 43(Database issue):D168-173.
[37]Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 ca talog of human long noncoding RNAs:analysis of their gene structure,evolution, and expression[J]. Genome Research, 2012, 22(9):1775-1789.
[38]Volders PJ, Helsens K, Wang X, et al. LNCipedia:a database for annotated human lncRNA transcript sequences and structures[J]. Nucleic Acids Research, 2013, 41(Database issue):D246-251.
[39]Bu D, Yu K, Sun S, et al. NONCODE v3. 0:integrative annotation of long noncoding RNAs[J]. Nucleic Acids Research, 2012, 40(Database issue):D210-215.
[40]Xie C, Yuan J, Li H, et al. NONCODEv4:ex ploring the world of long non-coding RNA genes[J]. Nucleic Acids Research, 2014,42(Database issue):D98-103.
[41]Chan WL, Huang HD, Chang JG. lncRNAMap:a map of putative regulat ory functions in the long non-coding transcriptome[J].Computational Biology and Chemistry, 2014, 50:41-49.
[42]Bhartiya D, Pal K, Ghosh S, et al. lncRNome:a com prehensive knowledgebase of human long noncoding RNAs[J]. Database :the Journal of Biological Databases and Curation, 2013, 2013:bat034.
[43]Niazi F, Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3’UTRs[J]. RNA, 2012, 18(4):825-843.
[44]Li JH, Liu S, Zhou H, et al. starBase v2. 0:decoding miRNA-ceRNA, m iRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data[J]. Nucleic Acids Research,2014, 42(Database issue):D92-97.
[45]Dinger ME, Pang KC, Mercer TR, et al. NRED :a database of long noncoding RNA expression[J]. Nucleic Acids Research, 2009,37(Database issue):D122-126.
[46]Gong J, Liu W, Zhang J, et al. lncRNASNP:a d atabase of SNPs in lncRNAs and their potential functions in human and mouse[J].Nucleic Acids Research, 2015, 43(Database issue):D181-186.
[47]Ding J, Lu Q, Ouyang Y, et al. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice[J]. Proceedings of the National Academy of Sciences of theUnited States of America, 2012, 109(7):2654-2659.
[48]Denman RB.Using RNAFOLD to predict the activity of small catalytic RNAs[J]. BioTechniques, 1993, 15(6):1090-1095.
[49]Andronesc u M, Aguirre-Hernandez R, Condon A, et al. RNAsoft:A suite of RNA secondary structure prediction and design software tools[J]. Nucleic Acids Research, 2003, 31(13):3416-3422.
[50]Zuker M. Mfold web server for nucleic acid folding and hybridization prediction[J]. Nucleic Acids Research, 2003, 31(13):3406-3415.
[51]Doench JG, Sharp PA. Specificity of microRNA target selection in translatio nal repression[J]. Genes &Development, 2004, 18(5):504-511.
[52]Barenboim M, Zoltick BJ, Guo Y, et al. MicroSNiPer:a web tool for prediction of SNP effe cts on putative microRNA targets[J].Human Mutation, 2010, 31(11):1223-1232.
[53]Sabarinathan R, Tafer H, Seemann SE, et al. The RNAsnp web server:predicting SNP effects on local RNA seco ndary structure[J]. Nucleic Acids Research, 2013, 41(Web Server issue):W475-479.
[54]Bruno AE, Li L, Kalabus JL, et al. miRdSNP:a database of disease-associated SNPs and microRNA target sites on 3’UTRs of human genes[J]. BMC Genomics, 2012, 13:44.
[55]Liu C, Zhang F, Li T, et al. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-relate d SNPs in GWAS SNPs and eQTLs[J]. BMC Genomics, 2012, 13:661.
[56]Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets[J]. Cell, 2005, 120(1):15-20.
[57]Paraskevopoulou MD, Georgakilas G, Kostoulas N, et al. DIANA-microT web server v 5. 0:service integration into miRNA functional analysis workflows[J]. Nucleic Acids Research, 2013,41(Web Server issue):W169-173.
[58]Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions[J]. Nature Genetics, 2005, 37(5):495-500.
[59]Rusinov V, Baev V, Minkov IN, et al. MicroInspector:a w eb tool for detection of miRNA binding sites in an RNA sequence[J].Nucleic Acids Research, 2005, 33(Web Server issue):W696-700.
[60]Gong J, Wu Y, Zhang X, et al. Comprehensive analysis of hum an small RNA sequencing data provides insights into expression profiles and miRNA editing[J]. RNA Biology, 2014, 11(11):1375-1385.
[61]Jin G, Sun J, Isaacs SD, et al. H uman polymorphisms at long noncoding RNAs(lncRNAs)and association with prostate cancer risk[J]. Carcinogenesis, 2011, 32(11):1655-1659.
[62]Ning S, Zhao Z, Ye J, et al. LincSNP:a database of linking disease-associated SNPs to human large intergenic non-coding RNAs[J]. BMC Bioinformatics, 2014, 15:152.
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
今日农业(2021年11期)2021-08-13 08:53:24
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:12
汉字汉语研究(2020年2期)2020-08-13 07:52:48
电子制作(2019年22期)2020-01-14 03:16:24
生物学通报(2019年3期)2019-02-17 18:03:58
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
