
几种含有二氟甲氧基桥键的新型液晶合成
2015-08-21 09:05:36闻建勋王建新杜宏军李继响
化工生产与技术 2015年1期
闻建勋 王建新 赵 敏 杜宏军 李继响
(1.上海天问化学有限公司,上海200232;2.华东理工大学化学与分子工程学院,上海200237;3.福建省邵武市永晶化工有限公司,福建 邵武354001)
在学术研究的进展及工业方面应用的成功,液晶化合物已经是引人瞩目的材料[1-4]。液晶平板显示是现在可携带电脑、手机、航海航空系统及电视机平板显示屏的主流。这些应用在响应速度及图像反差方面必须满足高要求。在液晶材料的物理性质方面,考虑到集成电路(IC)的耐电压,要求低的驱动电压,即要求使用阈值电压低的液晶及液晶组合物。一般的做法是使用介电常数各向异性Δε较大的液晶化合物,可以降低阈值电压Uth。因此,开发介电常数各向异性大的极性液晶的工作受到重视[3]。
由于氟原子在元素中电负性最高,因而将氟原子作为取代基导入液晶分子,可以增大Δε,利用增加氟原子数目的方法十分有效。但是,虽然分子中的氟原子数目增加使黏度减小,但是液晶的清亮点也随之降低,结果使液晶相温度范围变低。单靠增加氟原子提高Δε,会引起黏度的升高。利用增加氟原子,抑制黏度上升的同时,就难于避免液晶相温度的下降,单靠控制Δε的变化是非常困难的。
为了达到增加Δε而不使黏度过多增大的目的,德国MERCK公司的研究人员于1989年首先报道了CF2O桥键的新型化合物的合成[5]。但是当时数据不多,难于系统研究。到了20世纪90年代,这类化合物的合成逐渐增加,使它们的性质得到系统研究。研究结果发现有些CF2O化合物不仅黏度低,而且有良好的脂溶性[6-10]。1995年MERCK申请专利[11]之后,日本的CHISSO公司也申请专利。尽管这些化合物本身并不显示液晶相,但是作为组分加入液晶组合物,显著改善了后者的液晶性,人们也把它们视为液晶对待。
在液晶分子的2个苯环之间,导入桥键CF2O的化合物,近10年来多有报道。如果桥键CF2O的极化作用的贡献与分子长轴的极性是一致的,就可以得到比较大的Δε。2个氟原子保持在分子的桥键之中,实验发现黏度可以大大减小。
本工作开发了含有CF2O的含氟化合物,特点是分别利用了不同的末端环。尤其是3-氟-4-三氟甲基苯基团、1,3-二氧杂-5-烷基-2-环己基-,它们的极性方向与分子长轴一致,有助于Δε的增大。由于苯环上的CF3—的导入,不仅增大极性,而且非常有助于脂溶性的改善,避免低温时组分析出[10]。
在合成CF2O结构的工艺中,利用二氟二溴甲烷作原料的合成路线方法,工艺简单。
1 目标化合物的合成
1.1 合成路线
研究共合成4类含CF2O桥键的含氟液晶化合物,其结构如下:
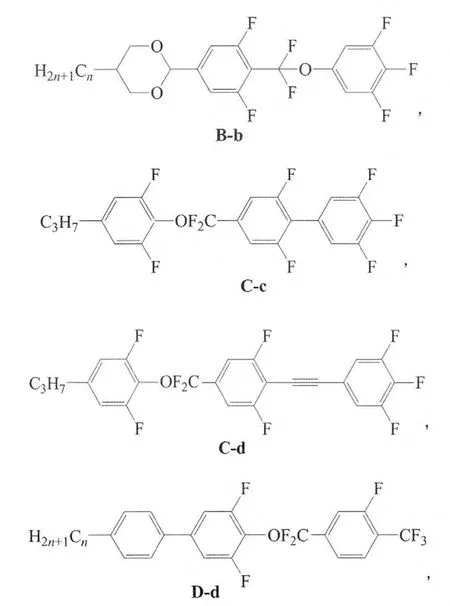
其中,n=1、2、3、4、5或6。
1)二氟-[(5-烷基-1,3-二氧杂-环己基)-2,6-二氟苯基]-(3,4,5-三氟苯氧基)甲烷(B-b)的合成:
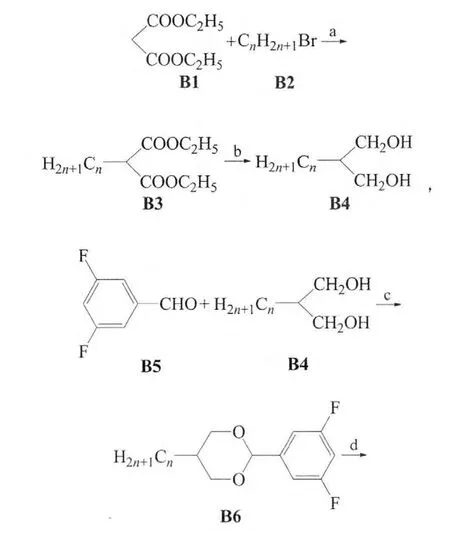

其中,n=1、2、3、4、5或6。
2)二氟-[4-正丙基-2,6-二氟苯基]-(3,5,3′,4′,5-五氟联苯氧基)甲烷的合成:
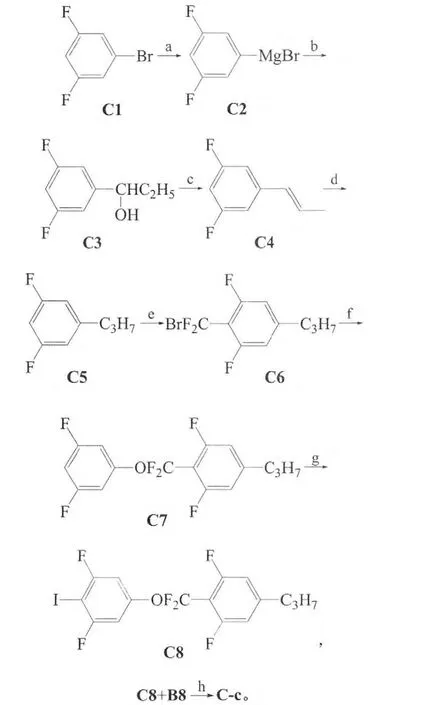
3)二氟-[4-正丙基-1,3-二氟苯基]-[(2,3,4,5,6-五氟苯基)乙炔基]-3,5-二氟苯氧基]甲烷的合成:

4)二氟-[4-正烷基苯基-2,6-二氟苯基]-(3-氟-4-三氟甲苯氧基)甲烷的合成(D-d):
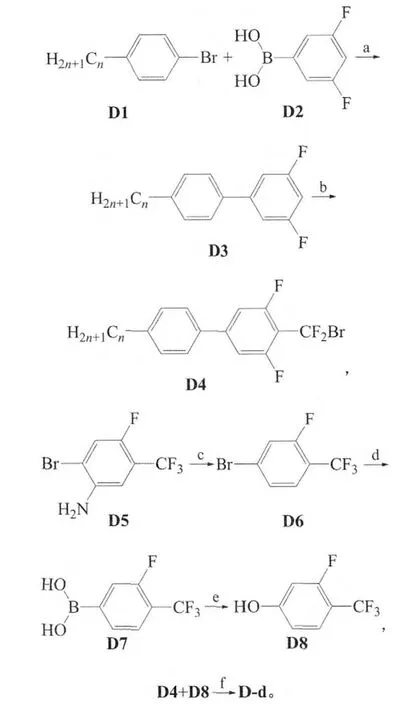
其中,n=1、2、3、4、5或6。
1.2 实验方法
实验仪器:Bruker 400(400 MHz)型核磁共振仪测,G2577A型质谱仪,Nicolet Magna-I 550型红外光谱仪,XPV-203E型偏光显微镜,Dimand DSC型差示扫描量热仪。
1.2.1 二氟-[(5-烷基-1,3-二噁烷基)-2,6-二氟苯基]-(3,4,5-三氟苯氧基)甲烷(B-b,n=5)的合成
1)2-戊基丙二酸二乙酯的合成。在干燥的500 mL三口烧瓶中加入钠片11.5 g及无水乙醇250 mL,装上回流冷凝管和滴液漏斗。待钠片反应完毕,滴加丙二酸二乙酯80.0 g,然后再滴加正戊基溴83.0 g,之后回流0.5 h。加醋酸中和后,过滤除去溴化钠固体。除去滤液中的溶剂,加入稀盐酸,用甲基叔丁基醚萃取。用无水硫酸镁干燥,减压除去溶剂,所得物减压蒸馏(82~84℃/133 Pa)得无色液体70.0 g。粗产率61%。
2)2-戊基-丙-1,3-二醇(C5H11CH(CH2OH)2)的合成。在干燥的500 mL三口烧瓶中装上回流冷凝管和滴液漏斗,氮气保护下加入LiAlH4的15.0 g、四氢呋喃(THF)250 mL,然后慢慢滴加2-戊基丙二酸二乙酯40.0 g。滴加完毕,回流1 h后,冷却,慢慢滴加15mL水,再慢慢滴加质量分数10%的NaOH水溶液,直到固体全部变成白色。过滤,滤液用无水硫酸镁干燥。减压除去溶剂,所得物减压蒸馏得无色液体15.0 g,粗产率59%。
3)3,5-二氟苯甲醛的合成。在干燥的500mL三口烧瓶中加入镁屑10.0 g及THF 300mL,然后慢慢滴加3,5-二氟溴苯77.2 g,制成格氏试剂。冷却至0℃,慢慢滴加N,N-二甲基甲酰胺(DMF)32mL,然后让其自然升至室温,反应4 h。加水后用甲基叔丁基醚萃取,无水硫酸镁干燥。减压除去溶剂,所得物减压蒸馏得白色液体41.0 g,粗产率72%。
4)2-(3,5-二氟苯基)-5-戊基-1,3-二噁烷的合成。在500mL单口瓶中加入3,5-二氟苯甲醛4.0 g、对甲苯磺酸0.5 g、甲苯150 mL、2-戊基-丙-1,3-二醇4.6 g,然后回流分水。当不再有水出来时(约3 h),停止加热。冷却后加入K2CO32.5 g,搅拌10 min。水洗后,用无水硫酸镁干燥。减压除去溶剂,用柱层析分离(石油醚和二氯甲烷体积比9:1),得固体石油醚重结晶得产物4.6 g,产率61%。MS((m/z)/%):270.1(M+,100.00),141.0(61.29);1H NMR(400 MHz,CDCl3)δ:0.81~1.54(m,11H),2.25(m,1H),4.02(m,2H),4.04~4.30(m,2H),5.34(s,1H),6.97~7.04(m,3H)。
其他同系物的合成方法与此相同,只是变更原料。其中2-(3,5-二氟苯)基-5-丙基-1,3-二烷,MS((m/z)/%):242.1(M+,100.00),141.0(61.29)。
5)2-(4-二氟溴甲基-3,5-二氟苯基)-5-戊基-1,3-二噁烷的合成。在100mL三口烧瓶中,装上滴液漏斗和低温温度计,氮气保护下加入2-(3,5-二氟苯基)-5-戊基-1,3-二噁烷2.2 g,干燥THF40mL。用丙酮-液氮冷却至-70℃,滴加正丁基锂4 mL后,控制温度-70℃左右反应2 h,再滴加CF2Br22.0 g。滴完后,控制温度在-70℃左右反应。反应2 h后停止反应,自然升至室温,加20mL水,用甲基叔丁基醚萃取,无水硫酸镁干燥。减压除去溶剂,用石油醚过柱,用石油醚重结晶得产物2.0 g,粗产率65%。MS((m/z)/%):397.0(M+,5.54),319.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.83~1.55(m,11H),2.35(m,1H),4.12(m,2H),4.14~4.40(m,2H),5.14(s,1H),6.97~7.05(m,2H)。
变更原料2-(3,5-二氟苯)基-5-丙基-1,3-二噁烷,采用相同的合成方法获得同系物2-(4-二氟溴甲基-3,5-二氟苯基)-5-丙基-1,3-二噁烷。MS((m/z)/%):369.0(M+,5.67),291.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.83~1.55(m,7H),2.35(m,1H),4.12(m,2H),4.14~4.40(m,2H),5.14(s,1H),6.97~7.05(m,2H)。
6)目标化合物二氟-[(5-烷基-1,3-二噁烷基)-2,6-二氟苯基]-(3,4,5-三氟苯氧基)甲烷(n=5)的合成。在100mL三口烧瓶中加入2-(4-二氟溴甲基-3,5-二氟苯基)-5-戊基-1,3-二噁烷2.0 g、3,4,5-三氟苯酚0.8 g、四丁基溴化铵1.0 g、碳酸钾2.5 g及DMF 50 mL,加上回流冷凝管,在90~100℃反应3 h。冷却,加入40mL水搅拌,将固体溶解。用甲基叔丁基醚萃取,用有机相水洗,无水硫酸镁干燥。减压除去溶剂,用石油醚过柱,甲醇重结晶2次,得白色晶体0.5 g,产率29%。相变温度/℃:Cr 41.67 I 10.32 Recr(Recr代表冷却过程时成为晶体)。MS((m/z)/%):466.1(M+,4.39),319.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.89(t,J=6.4 Hz,3H),1.29~1.31(m,6H),1.55(s,2H),2.08~2.11(m,1H)4.12(m,2H),3.49~4.25(m,4H),5.36(m,1H),6.93~6.96(m,2H),7.12~7.15(m,2H)。
目标化合物二氟-[(5-烷基-1,3-二噁烷基)-2,6-二氟苯基]-(3,4,5-三氟苯氧基)甲烷(n=3)的相同方法。MS((m/z)/%):438.1(M+,0.98),291.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.88(t,J=6.4 Hz,3H),1.29~1.33(m,2H),1.54(s,2H),2.09~2.12(m,1H),4.13(m,2H),3.48~4.24(m,4H),5.37(m,1H),6.92~6.967(m,2H),7.11~7.16(m,2H)。
1.2.2 二氟-[4-正丙基-2,6-二氟苯基]-(3,5,3′,4′,5-五氟联苯氧基)甲烷(C-c)系列的合成
1)C3。将2.5 g镁和20 mL干燥的THF,在氮气保护下加入250mL的三口烧瓶中。磁力搅拌下,再将19.3 g的3,5-二氟溴苯溶于70 mL干燥的THF中并置于100mL的常压滴液漏斗中。滴入少量混合液(10 mL)使镁屑激活,5 min后升温并回流,继续滴加剩余的混合液。40min滴完后,继续在常温反应1.5 h,滴加丙醛5.8 g和60mL乙醚的混合液,10min滴完,加热回流2 h。冷却,慢慢加入250mL饱和的氯化铵溶液(过快会爆沸),先会有沉淀生成,继续滴加沉淀溶解。用甲基叔丁基醚提取混合液2次,得有机相再用水洗2次,干燥,然后用旋转蒸发仪蒸馏干,过柱(石油醚与乙醚体积比20:1),得到淡黄色液体13.4 g。粗产率78%。MS((m/z)/%):172.1(M+,12.41),143.0(100.00);1H NMR(400MHz,CDCl3)δ:0.82(t,J=7.2 Hz,3H),1.60~1.68(m,2H),2.23(s,1H),4.48(t,J=6.4 Hz,1H),6.58~6.79(m,3H)。
2)C5。将130 mL的正己烷和28.4 g五氧化二磷加入250mL的三口烧瓶中,机械搅拌,慢慢加入13.4 g的1-(3,5-二氟苯基)-丙基-1-醇,加热至50~60℃反应,2.5 h后,点板反应完全,抽滤,得母液。将母液加入反应釜中,加催化量的Pd/C,在3 MP的氢气压力下常温搅拌过夜。气相跟踪,反应完全,抽滤,得母液旋干,减压蒸馏收集58~60℃/2.66 kP的馏分6.0 g,产率50%。
3)C6。将4.0 g的3,5-二氟丙苯和70 mL干燥的THF在氮气保护下加入100 mL的三口烧瓶中,降温至-78℃以下滴加12 mL的正丁基锂,控制温度在-70℃以下,20min滴完,继续低温反应2 h,滴加CF2Br28 mL,滴加控制温度在-70℃以下,5 min滴完,继续反应2 h,气相跟踪,反应完全,停止,自动升至常温,加入水30 mL,用甲基叔丁基醚提取有机相2次,干燥,旋干,用石油醚过柱,得产品6.3 g,产率86%。
4)C7。将4.5 g的2-(溴-二氟甲基)-1,3-二氟-5-丙基苯、2.3 g的3,5-二氟苯酚、1.1 g的四丁基溴化铵、4.5 g的碳酸钾和80mL的DMF,加入100mL的三口烧瓶中。磁力搅拌下,加热至100℃左右,反应3 h。用点板法待反应完全后,冷却,加入60 mL水,搅拌使固体溶解。用甲基叔丁基醚提取有机相3次,合并有机相,再用水洗1次。得有机相干燥,旋转蒸馏去溶剂,用石油醚过柱,得到无色液体3.0 g,粗产率:57%。MS((m/z)/%):334.1(M+,2.42),205.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.95(t,J=7.2 Hz,3H),1.62~1.68(m,2H),2.59(t,J=7.6 H,2H),6.68~6.85(m,5H)。
5)C8。在氮气保护下,将0.4g的二异丙胺和30 mL的THF加入100 mL的三口烧瓶中。降温至-5℃以下,滴加正丁基锂,滴完后继续低温反应1 h,放置待用。将1.0 g的2-[(3,5-二氟苯羟基)-二氟甲基]-1,3-二氟-5-丙基苯和30 mL干燥的THF加入到100mL三口烧瓶中,氮气保护。降温至-78℃以下,滴加二异丙基胺基锂(LDA)。10min滴完,继续低温反应2 h,自动升温至-60℃,滴加单质碘,滴完后继续在-60℃反应1.5 h,停止反应。自动升温至常温,加入50 mL饱和氯化铵溶液。分层,无机层用乙酸乙酯洗3次,合并有机相,再用硫代硫酸钠水溶液洗3次,得有机相,干燥。旋转蒸馏除去溶剂。用石油醚过柱,得到产品0.98 g,粗产率70%。MS((m/z)/%):460.0(M+,12.98),205.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.95(t,J=7.2 Hz,3H),1.62~1.68(m,2H),2.59(t,J=7.6 Hz,2H),6.79~6.90(m,4H)。
6)目标化合物二氟-[4-正丙基-2,6-二氟苯基]-(3,5,3′,4′,5′-五氟联苯氧基)甲烷。用搅拌使原料溶解,加入化合物C9 0.34 g,催化量的Pd(PPh3)4与碳酸钾。加热回流条件下反应5 h,停止反应。反应液冷却至常温,抽滤,得到的母液用甲基叔丁基醚和水混合萃取,得到有机相,用水洗1次,干燥,浓缩,石油醚过柱得目标化合物0.57 g,产率71%。MS((m/z)/%):464.1(M+,11.03),205.1(100.00);1H NMR(400 MHz,CDCl3)δ:0.96(t,J=7.2Hz,3H),1.64~1.69(m,2H),2.61(t,J=7.2Hz,2H),6.81~6.84(m,2H),6.97~6.99(m,2H),7.08~7.12(m,2H)。
1.2.3 化合物二氟-[4-正丙基-1,3-二氟苯基]-[(2,3,4,5,6-五氟苯基)乙炔基]-3,5-二氟苯氧基]甲烷系列的合成
在100 mL干燥的三口烧瓶中加入化合物C8 0.8 g,加入溶剂三乙胺60 mL,搅拌使原料溶解,加入化合物C10 0.37 g,催化量的Pd(PPh3)2Cl2与CuI。加热回流条件下反应5 h,停止反应。反应液冷却至常温,抽滤,得母液用甲基叔丁基醚和水混合萃取,得到有机相水洗1次,干燥、浓缩,石油醚过柱得目标化合物0.55 g,产率60%。相变温度/℃:Cr 46.4 I 15.5 Recr;MS((m/z)/%):524.1(M+,12.58),205.1(100.00);1H NMR(400MHz,CDCl3)δ:0.95(t,J=7.2Hz,3H),1.61~1.68(m,2H),2.61(t,J=8.0Hz,2H),6.80~6.83(m,2H),6.93~6.96(m,2H)。1.2.4化合物二氟-[4-正烷基苯基-2,6-二氟苯基]-(3-氟-4-三氟甲苯氧基)甲烷系列的合成(D-d)
1)D3(n=5)。在氮气保护下向干燥的250 mL三口烧瓶中加化合物D1(n=5)10.0 g和化合物D2 7.7 g,加干燥的无水乙醇150 mL搅拌使溶解。加入碳酸钾5.0 g,催化剂Pd(PPh3)41.0 g,回流条件下反应5 h,停止反应。反应液抽滤,滤饼甲基叔丁基醚洗涤2次,加少量水,甲基叔丁基醚萃取,合并有机相,用无水硫酸镁干燥。抽滤,旋转蒸发仪除去溶剂,石油醚过柱得白色固体8.0 g,产率70%。MS((m/z)/%):260.1(M+,31.54),203.1(100.00)。
同系物合成方法相同,D3(n=3)。MS((m/z)/%):232.1(M+,31.54),203.1(100.00)。
2)D4(n=5)。在氮气保护下向干燥的100mL三口烧瓶中加化合物D3(n=5)5.0 g和干燥的THF 40 mL,用液氮-丙酮使溶液温度降低至-70℃,滴加含正丁基锂7.7 mL(2.5 mol/L)的30 mLTHF混合液,控制反应液温度在70℃左右,滴加完毕后在70℃条件下反应2.5 h。滴加二氟二溴甲烷4.4 g,控制温度于70℃左右,滴加完毕反应2 h。加入少量氯化铵饱和溶液搅拌5 min,自然升至室温,向反应液中少量水,甲基叔丁基醚萃取3次,合并有机相,无水硫酸钠干燥,过滤,减压除去溶剂,石油醚过柱得白色固体D4(n=5)4.5 g,粗产率61%。1H NMR(400 MHz,CDCl3)δ:0.88(t,J=7.2 Hz,3H),1.45~1.64(m,6H),2.55(t,J=7.6 Hz,2H),6.98~7.03(m,2H),7.15~7.18(m,2H),7.36~7.38(m,2H)。
同系物合成方法相同。其中D4(n=3),1H NMR(400 MHz,CDCl3)δ:0.88(t,J=7.2 Hz,3H),1.45~1.64(m,2H),2.55(t,J=7.6 Hz,2H),6.98~7.03(m,2H),7.15~7.18(m,2H),7.36~7.38(m,2H)。
3)D6。称取75.0 g的D5,加入1 000mL干燥的三口烧瓶中。将278mL浓HCl与278mL水配成的溶液于搅拌下滴入,15min滴完,水浴加热,控制反应温度在60℃条件下反应4 h。停止加热,用冰水混合物使反应液温度降至0℃左右,滴加21.0 g的NaNO2和50 mL的水配成的溶液,25 min滴完,反应液由白色转变为淡黄色,继续低温反应1 h。称92.4 g的H3PO2于100 mL常压滴液漏斗中滴加入反应液中,25 min滴加完,此时反应液温度为15℃,再继续反应4 h,停止反应。水泵抽滤,母液用盐水萃取2次,得到有机相用无水硫酸镁干燥,减压蒸馏得到50.0 g无色液体(66~68℃/3.72 kPa)。石油醚过柱得无色液体45.3 g,粗产率64%。MS((m/z)/%):243.9(M+,99.67),241.9(100.00);1HNMR(400 MHz,CDCl3)δ:7.41(d,J=9.2Hz,2H),7.48(t,J=8.0 Hz,1H)。
4)3-氟-4-三氟甲基苯硼酸。在氮气保护下将6.0g的化合物D6和10mL干燥的THF加入100 mL干燥的三口烧瓶中,降温到-20℃,滴加35mL i-PrMgCl/THF,10min滴完,维持相同温度反应2 h,滴加5.3g硼酸三异丙酯,5min滴完,继续低温反应2h。滴加3.6g质量分数30%的盐酸,-10℃下反应1h。之后自然升至室温,减压蒸除THF,用乙酸乙酯和水混合液溶解粗产物,分层,水相用乙酸乙酯萃取3次,合并有机相,用无水硫酸钠干燥。减压蒸除溶剂,得固体用正己烷打浆,冷却后,静置,抽滤,得白色固体产物3.1g。产率60%。MS((m/z)/%):208.0(M+,94.32),144.0(100.00);1HNMR(400MHz,氘 代DMSO)δ:7.74~7.78(m,3H),8.56(s,2H);IR(KBr,νmax/cm-1):3390、3316、1630、1574、1515、1415、1319、1138、1043、904、841、812、684、608。
5)D8。将2.1g3-氟-4-三氟甲基苯硼酸加入100mL三口烧瓶中,加入冰醋酸3.0g和80mL的THF使之溶解。磁力搅拌下,将24.1g(质量分数30%)双氧水从滴液漏斗中慢慢滴入。水浴控制温度低于40℃,待温度稳定后常温反应24h,停止反应。加质量分数15%的硫酸钠溶液和甲基叔丁基醚混合萃取3次,合并有机相,干燥,浓缩后过柱(石油醚与二氯甲烷体积比1:1),得淡黄色液体1.3g,产率71%。MS((m/z)/%):180.0(M+,100.00),161.0(78.33),130.0(33.37);1HNMR(400MHz,CDCl3)δ:3.74~3.77(m,1H),6.53~6.57(m,3H)。
6)目标化合物二氟-[4-正烷基苯基-2,6-二氟苯基]-(3-氟-4-三氟甲苯氧基)甲烷(n=5)。在一干燥的100mL三口烧瓶中加化合物D4(n=5)2.0g和化合物D81.0g,加70mL的DMF搅拌使其溶解,加无水碳酸钾1.0g和四丁基溴化铵1.0g。搅拌并加热控制温度在90~100℃反应4h,停止反应,冷却后加水使固体溶解。甲基叔丁基醚萃取3次,合并有机相,无水硫酸钠干燥。抽滤,减压除去溶剂,石油醚过柱,甲醇重结晶得最终白色晶体1.6g。产率64%。MS((m/z)/%):460.1(M+,1.98),281.1(100.00);1HNMR(400MHz,CDCl3)δ:0.97(t,J=6.8Hz,3H),1.66~1.71(m,6H),2.65(m,2H),7.18~7.64(m,9H)。
目标化合物二氟-[4-正烷基苯基-2,6-二氟苯基]-(3-氟-4-三氟甲苯氧基)甲烷(n=3),MS((m/z)/%):488.1(M+,1.83),309.1(100.00);1HNMR(400 MHz,CDCl3)δ:0.98(t,J=6.8Hz,3H),1.67~1.73(m,2H),2.66(m,2H),7.19~7.66(m,9H)。
2 相变及性质研究
2.1 B-b系列化合物
B-b系列并没有出现液晶相,该化合物直接由晶体转化为各向同性的液体。B-bn=5时其熔点为41.7℃,n=3时熔点为40.2℃。化合物B-bn=5的DSC如图1。
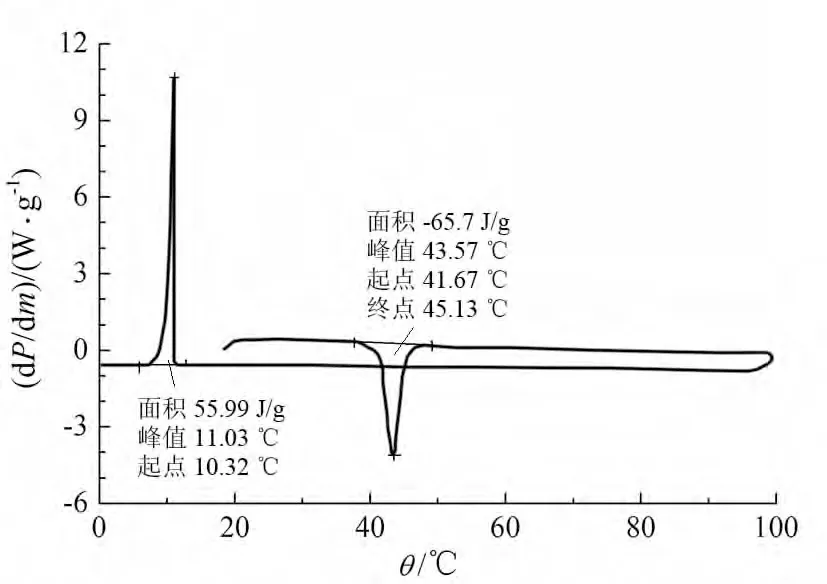
图1 化合物B-b(n=5)的DSC Fig 1 The DSC of compound B-b(n=5)
化合物B-b系列的分子相变温度见表1。
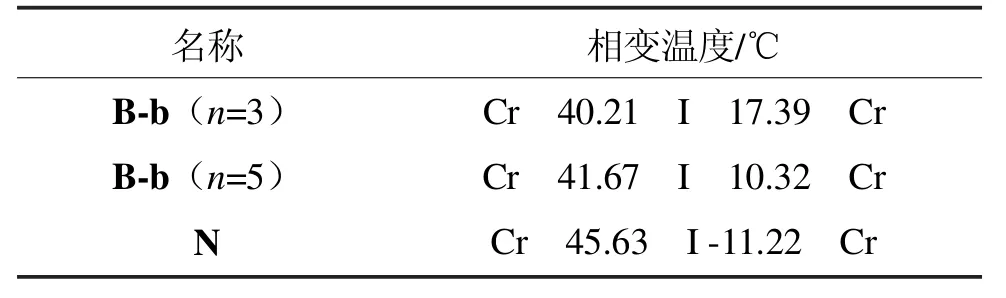
表1 化合物B-b的性质及相变Tab 1 Phase transition temperature and property of compounds B-b
从表1可以看出,化合物N是文献报道的化合物{3},目标化合物B-b与N的区别在于用其中1,3-二噁烷代替了苯环,2个氧原子形成的偶极矩与氟原子的一致,其Δε有所增加。
2.2 其他系列
从化合物C-c、C-d、D-d的分子结构可以看出,3类化合物侧面有较多氟原子取代基,分子间引力大大减少,不足以形成液晶相。3类化合物C-c、Cd、D-d设计是为了利用其高的Δε来提高混晶配方Δε,而且该类化合物的脂溶性良好,可以很好保持组合物的低温稳定性。
3 结论
利用二溴二氟甲烷与丁基锂反应的路线,与早期文献的硫化物氟化方法比较,工艺简便可行[3]。可以预期本研究的化合物较文献的化合物N有较大的Δε,在TFT组合物的开发上有应用价值。
[1]Hird M.Fluorinated liquid crystals-propertiesand applications[J].Chem Soc Rev,2007,36:2070-2095.
[2]Kirsch P,Bremer M.Nematische flüssigkristalle für aktivmatrix-displays:design und synthese[J].Angew Chem,2000,112:4384-4405.
[3]Pauluth D,Tarumi K.Advanced liquid crystals for television[J].JMater Chem,2004,14:1219-1227.
[4]Pauluth D,Tarumi K.Optimization of liquid crystals for television[J].JSID,2005,13:693-702.
[5]Bartmann E,Hittich R,Kurmeier A,etal.Difluoro-methylene cpds.-useful as low-viscosity components of liq.crystalline media for liq.crystal and electro=optical display elements.DE,4006921[P].1990-09-20.
[6]Kirsch P,BremerM,Taugerbeck A,etal.Difluorooxymethylene-bridged liquid crystals:a novel synthesis based on the oxidative alkoxydifluorodesulfuration of dithianylium salts[J].Angew Chem,2001,113:1528-1532.
[7]Kirsch P,BremerA,Taugerrbeck A,etal.Difluorooxymethylene-bridged liquid crystals:a novel synthesis based on the oxidative alkoxydifluorodesulfuration of dithianylium Salts[J].Angew Chem,Int Ed,2001,40:1480-1484.
[8]Kirsch P,Bremer M,Heckmeier M,et al.Flüssigkristalle auf der basis hypervalenter schwefelfluoride:pentafluorsulfuranyl als polare Endgruppe[J].Angew Chem,1999,111:2174-2178.
[9]P Kirsch,A Hahn.Liquid crystals based on hypervalent sulfur fluorides:exploring the steric effects ofortho-fluorine substituents[J].Eur JOrg Chem,2005:3095-3100.
[10]Smart B E.Fluorine substituent effects(on bioactivity)[J].J Fluorine Chem,2001,109:3-11.
[11]T Kazuaki,B Ekkehard.New alpha,alpha-di:fluorobenzyletherderivs:DE,19531165[P].1996-03-07.
猜你喜欢
云南化工(2021年5期)2021-12-21 07:41:12
农药科学与管理(2019年8期)2019-11-23 08:04:44
益寿宝典(2017年1期)2017-09-03 10:30:27
中小学实验与装备(2016年1期)2016-04-19 00:02:28
化学工业与工程(2015年1期)2015-02-10 03:01:33
红领巾·探索(2014年8期)2014-10-10 02:14:02
应用化工(2014年3期)2014-08-16 13:23:50
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年7期)2014-02-28 17:32:28
应用技术学报(2014年1期)2014-02-28 14:52:13
