
芳乙酸类化合物的合成研究进展I
2015-12-17 08:37叶盼盼金耀来郑土才潘向军郭宇欣
化工生产与技术 2015年1期
叶盼盼 金耀来 郑土才* 潘向军 郭宇欣 王 吉
(1.衢州学院化学与材料工程学院,浙江 衢州 324000;
2.江西吉翔医药化工有限公司,江西乐平333300)
芳乙酸类化合物的合成研究进展I
叶盼盼1金耀来1郑土才1*潘向军1郭宇欣1王 吉2
(1.衢州学院化学与材料工程学院,浙江 衢州 324000;
2.江西吉翔医药化工有限公司,江西乐平333300)
介绍了芳乙酸类化合物的特性,总结了零碳取代芳烃与两碳原子及以上单元直接或间接合成芳乙酸的方法,具体按照芳烃与2个碳原子单元反应法、芳胺经吲哚酮或吲哚醌合成法、卤代芳烃一步引入2个及以上碳原子的合成法合成芳乙酸进行讨论,叙述了这些方法的特点及适用范围。
芳乙酸;草酰氯单酯;扁桃酸;吲哚酮;格氏偶联;丙二酸酯;亲核取代;合成
芳乙酸是医药、农药等产品的常见结构或结构单元,如非甾体抗炎药联苯乙酸、双氯芬酸和罗美昔布,植物生长调节剂α-萘乙酸和3-吲哚乙酸[1-4]。许多芳乙酸是重要的医药、农药、液晶材料等中间体[5]。如苯乙酸为青霉素G发酵过程的重要添加剂,也是合成异黄酮的原料,对羟基苯乙酸是降压药阿替洛尔和杀虫剂乙氰菊酯的中间体,2,4,5-三氟苯乙酸是新型抗糖尿病药西他列汀的关键中间体,对三氟甲基苯乙酸为新药中间体,3-乙氧基-4-乙氧羰基苯乙酸是抗糖尿病药瑞格列奈的中间体,2-噻吩乙酸是广谱头孢菌素头孢噻吩、头孢噻啶的中间体,3-吡啶乙酸是抗骨质疏松药利塞膦酸钠的中间体,2,4-二氯苯乙酸、2,4,6-三甲基苯乙酸和2,5-二甲基苯乙酸则分别是杀虫剂螺螨酯、螺虫酯和螺虫乙酯的中间体,2,3-二氟苯乙酸为液晶材料中间体[6-14]。芳乙酸的衍生物还有许多其他应用,如合成γ-内分泌酶抑制剂,作为不对称亚胺配体和多核混合金属配合物的端位单元,合成手性药物左旋米那普仑,合成雌激素受体部分激动剂,作为钙(II)配位聚合物的配体,合成3,5-二芳基环戊烯酮和3-芳基-2-酰氧基丙烯酰胺等[15-21]。
根据C—C键的构建方法及芳烃取代基含碳个数,芳乙酸的合成有零碳取代芳环与2个碳原子及以上单元的直接或间接连接法、单碳取代芳环与单碳原子单元的连接法、单碳取代芳环与双碳原子单元的间接连接法、双碳取代芳环的取代基修饰法、以及芳乙酸的相互转化法,本文叙述第I部分——零碳取代芳环与2个碳原子及以上单元的直接或间接连接法合成芳乙酸。
1 零碳取代芳环与2个及以上碳原子单元连接法
1.1 芳烃与2个碳原子单元反应
1.1.1 芳烃与卤乙酸或卤乙腈的直接反应法
芳烃与氯乙酸、氯乙腈等在一定条件下可直接发生傅克烃基化反应,得到芳乙酸或芳乙腈,但该法的研究很少,适用范围也不很明确。
马冰洁等以萘和氯乙酸为原料,在纳米级复合金属混合物Fht为主催化剂,KBr为助催化剂作用下,直接合成植物生长调节剂α-萘乙酸[22]:
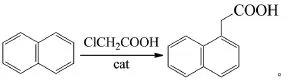
并探讨了原料配比、催化剂、反应时间等因素的影响,得到优化反应条件:萘与氯乙酸摩尔比为3:1,催化剂Fht与KBr质量比1:3,Fht-1用量为萘的质量0.5%,4 h升至反应温度180~224℃,反应时间
25 h,在该条件下产品收率61.4%。
柴多里等报道了类似工艺,但以铝粉为催化剂,优化反应条件为:萘与氯乙酸摩尔比1.5,铝粉用量为萘质量的2.6%,3 h升至反应温度185~208℃,反应时间20 h,在该条件下收率38.1%[23]。
张鹏翔等报道了1,2,4-三氟苯与氯乙腈直接傅克烃基化、水解制备2,4,5-三氟苯乙酸的2步法工艺[8]:
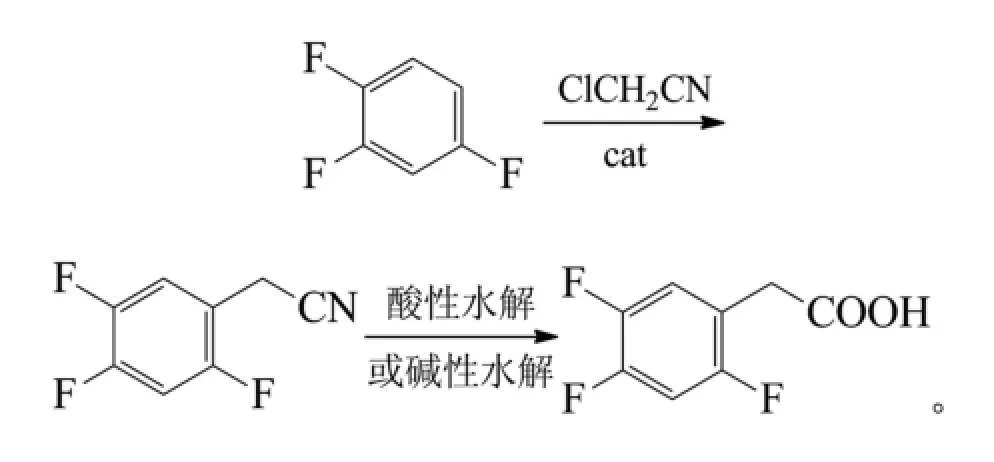
1,2,4-三氟苯与氯乙腈在路易斯酸如三氯化铁、三氯化铝、三氟化硼、四氯化锡、氯化锌等催化和溶剂如二氯甲烷、三氯甲烷、二氯乙烷等反应中得到2,4,5-三氟苯乙腈,收率72%~79%。水解可按常规方法进行,收率在90%以上。
1.1.2 芳烃与草酰氯单酯的傅克酰化、还原反应法
芳烃与草酰氯单乙酯经傅克酰化得到芳乙酮酸乙酯,再经酮羰基的Wolff-Kishner-黄鸣龙还原或Clemmensen还原及水解,是制备芳乙酸的较好方法,但酰化剂价格较高,限制了其应用。
陈刚等以苯甲醚为原料,在三氯化铝催化下与草酰氯单乙酯发生傅克酰化得到对甲氧基苯乙酮酸乙酯,收率85.5%。再在KOH作用下直接与水合肼发生还原反应合成药物中间体对甲氧基苯乙酸,产率为64.4%[24]:

姚国新等以邻苯二甲醚为原料,与草酰氯单乙酯在无水三氯化铝催化下生成3,4-二甲氧基苯乙酮酸乙酯,收率87.3%;再与水合肼发生还原反应后水解酸化得到中间体3,4-二甲氧基苯乙酸,收率73.4%[25]。
崔庆荣以噻吩为原料,经草酸单乙酯酰氯酰化反应得噻吩草酰乙酯,收率85.9%。再经水合肼-KOH还原水解生成2-噻吩乙酸,收率85%[26]。
向纪明等研究了芳烃与草酰氯单乙酯的傅克酰化反应,8例收率48%~82%。二氯甲烷或过量芳烃为较佳反应溶剂,方法具有条件温和、操作简单、收率高等优点。另外,高效液相色谱(HPLC)检测反应选择性发现,甲苯、乙苯、异丙苯、苯甲醚酰化时,虽然活性较高,但邻位异构体较多,达14.3%~29.3%。而弱吸电子基团取代的氟苯、氯苯和溴苯反应产物中邻位异构体很少,仅0.7%~1.3%[27]。
Ianni等在生物活性2-芳基取代肉桂酸酯类化合物的合成研究中,报道芳烃经草酰氯单乙酯的傅克酰化、酮羰基还原成醇、羟基的溴化及与亚磷酸三乙酯的Wittig-Horner反应得到2-芳基取代磷酰乙酸酯类中间体。傅克酰化以二氯甲烷为溶剂,三氯化铝为催化剂,6例收率77%~99%,芳烃包括邻苯二甲醚、对苯二甲醚、1,2,3-三甲氧基苯、苯甲醚、3,4-亚甲二氧基苯和1,4-苯并二噁烷[28]。
1.1.3 活泼芳烃与乙醛酸缩合、还原反应法
芳烃与乙醛酸缩合生成扁桃酸,再经直接或间接脱苄位羟基得到芳乙酸的方法,主要适用于带1个或以上羟基的高电子密度芳烃如苯酚。
乔喜龙等以乙醛酸和苯酚为原料,在NaOH作用下得到对羟基扁桃酸钠,亚磷酸还原得到对羟基苯乙酸。优化工艺条件:合成对羟基扁桃酸盐,n(乙醛酸):n(苯酚)=1:2,反应温度为(70±5)℃,反应时间为5 h,平均收率73.7%;合成对羟基苯乙酸,n(对羟基扁桃酸):n(亚磷酸)=1:1.05,反应温度为105℃,反应时间为6 h,平均收率为81.5%[29]。反应式为:
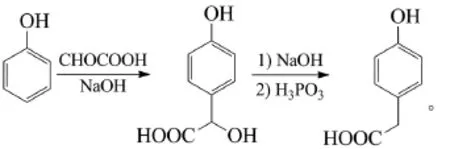
李胜辉等以乙醛酸和苯酚为起始原料,在NaOH中经缩合得到对羟基扁桃酸钠,收率85%。进一步经亚硫酸氢钠还原得到对羟基苯乙酸,收率86%。优化工艺条件:缩合反应,乙醛酸、苯酚、氢氧化钠的摩尔比为1:1.5:2.5;还原反应,对羟基扁桃酸钠、亚硫酸氢钠的摩尔比为1:1.1,反应温度100℃[30]。
张友恭等以乙醛酸和苯酚为原料,在碱性溶液中经缩合反应得到对羟基扁桃酸钠。再加焦亚硫酸钠进行还原得到对羟基苯乙酸,收率85%,优化工艺为:对羟基扁桃酸与焦亚硫酸钠摩尔比1:0.55,反应温度90~120℃,反应时间4~5 h[31]。
1.1.4 芳烃与二氯乙酰氯的傅克酰化、重排、还原反应法
芳烃与二氯乙酰氯傅克酰化,再经碱催化重排、水解生成扁桃酸,最后经与1.1.3节相似的脱苄位羟基反应,从而制得芳乙酸的方法,适用的芳烃范围比1.1.3节方法的更广,但反应步骤较多。
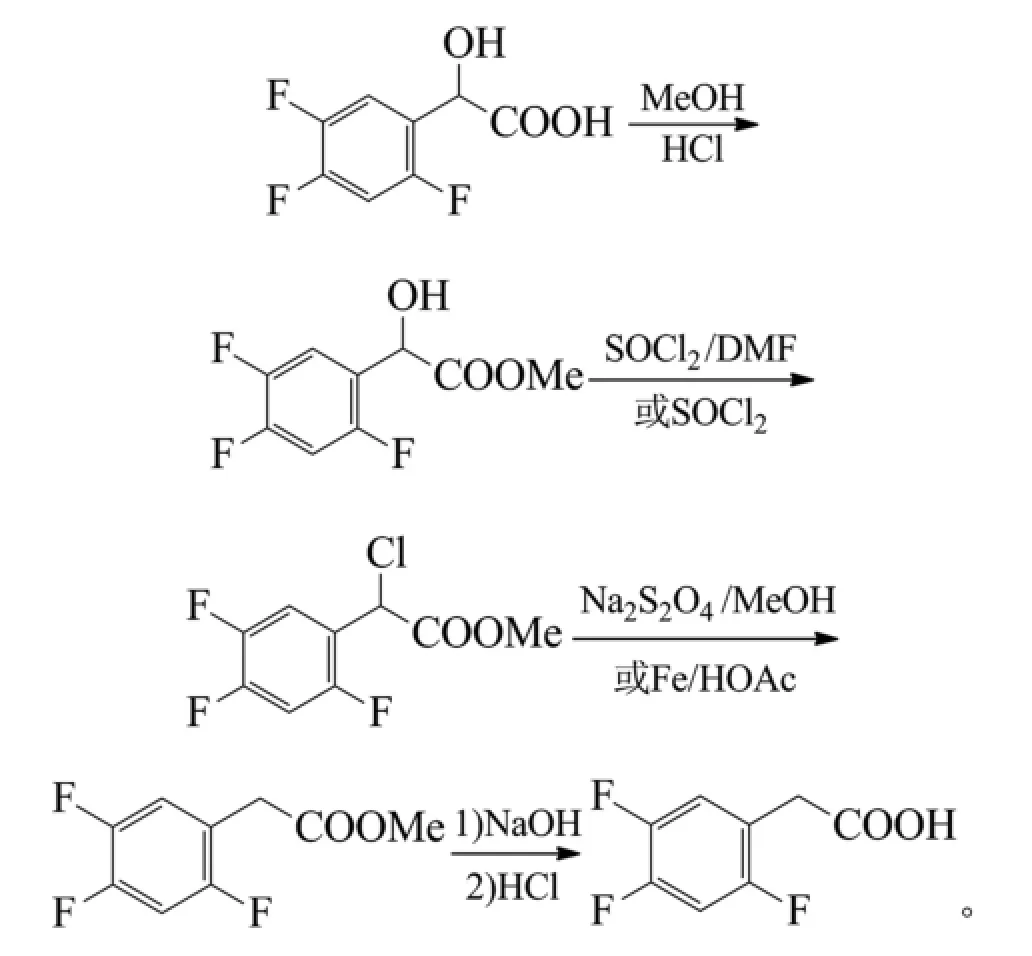
刘泽玲以1,2,4-三氟苯为原料,在三氯化铝、二氯乙酰氯作用下经傅克酰化得到α,α-二氯-2,4,5-三氟苯乙酮,收率95.1%。NaOH水解重排得到2,4,5-三氟扁桃酸,收率72.0%。再在SOCl2的二氯甲烷溶液中经氯代反应得到2-(2,4,5-三氟苯基)-2-氯乙酸,收率93.8%。最后在Pd/C催化下,在无水甲酸和三乙胺中,经甲酸脱氯得到2,4,5-三氟苯乙酸,收率56%[32]。反应式为:
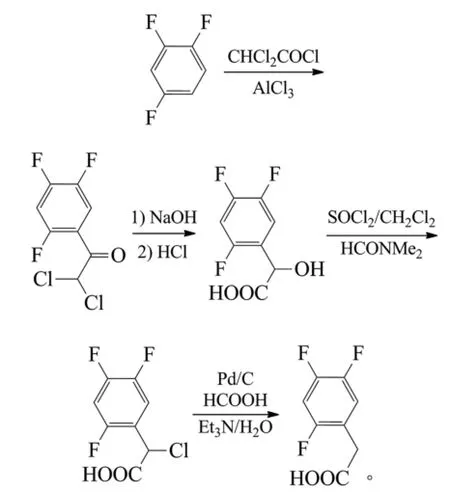
林艳艳等报道了1,2,4-三氟苯与二氯乙酰氯经傅克酰化制得2,4,5-三氟-α,α-二氯苯乙酮,碱催化重排、酸化制得2,4,5-三氟扁桃酸,再经氯化制得2,4,5-三氟-α-氯代苯乙酸,最后经还原脱氯制得2,4,5-三氟苯乙酸的工艺[33]。Falchi等以1,2,4-三氟苯与二氯乙酰氯/三氯化铝反应,经NaOH催化水解、重排、盐酸酸化得到2,4,5-三氟扁桃酸,2步收率84%。在氯化亚砜、N,N-二甲基甲酰胺(DMF)、二氯甲烷中直接氯化得到α-氯-2,4,5-三氟苯乙酸,收率90%,10%钯/炭催化下于甲酸、三乙胺、水中脱氯得2,4,5-三氟苯乙酸,收率83%[34]。
Falchi等还报道,2,4,5-三氟扁桃酸还可经质量分数3%的氯化氢甲醇进行甲酯化(收率94%)、氯化亚砜或氯化亚砜、DMF氯化得到α-氯-2,4,5-三氟苯乙酸甲酯,收率近乎定量。保险粉、甲醇或铁粉、醋酸还原脱氯得2,4,5-三氟苯乙酸甲酯,收率分别为73%和90%。NaOH水解、酸化得到2,4,5-三氟苯乙酸,收率95%[34]。反应式为:
1.2 芳胺经吲哚酮合成邻氨基芳乙酸
芳胺通过与氯乙酰氯的酰胺化或与草酰氯的单酰胺化,再在路易斯酸如三氯化铝催化下进行分子内傅克烃基化或分子内傅克酰基化和酮羰基还原生成吲哚酮,水解开环即得到邻氨基芳乙酸。根据需要,氨基可以保持不变,或重氮化转化为氢、卤素、氰基、羟基、巯基、肼基等。该方法反应步骤较多,主要用于用常规方法难以有效引入羧甲基的场合。
李桂英等以苯胺为原料,经氯乙酰氯化、聚乙二醇-600催化下与2,6-二氯苯酚醚化、NaOH存在下重排得到2,6-二氯二苯胺。该中间体与氯乙酰氯缩合、三氯化铝催化环合得1-(2,6-二氯苯基)-2-吲哚酮,2步收率90.5%。KOH水解得到非甾体抗炎药双氯芬酸钾,收率95%[35]。反应式为:

秦丙昌等以类似工艺合成了双氯芬酸钠,其中2,6-二氯二苯胺与氯乙酰氯的缩合及三氯化铝催化环合均在无溶剂条件下进行,最优条件为胺与氯乙酰氯摩尔比1:1.2,反应温度 140℃,缩合收率96.6%,三氯化铝用量2倍,反应温度150℃,环合收率91.2%,NaOH为2.5倍,质量分数20%,反应时间6 h,水解收率92.5%,3步总收率达81.5%[2]。魏文珑等也报道了类似工艺,优化了工艺条件,并经脱色、精制得精品双氯芬酸钠,3步收率分别为90.0%、82.1%和90.0%[36]。
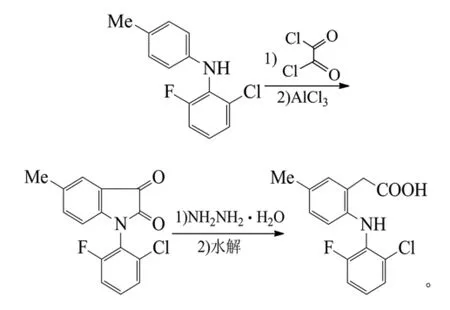
张爱华等以对甲苯胺和2-氯-6-氟苯酚为原料,经酰化、醚化、Smiles重排制得N-(2-氯-6-氟苯基)对甲苯胺,再经与草酰氯的单酰化、三氯化铝催化的分子内环合、酮羰基还原及水解开环得到新型COX-2抑制剂类抗炎药罗美昔布。其中单酰化以苯为溶剂,收率84%,环合以1,1,2,2-四氯乙烷为溶剂,收率86%,85%水合肼、KOH和乙二醇还原、酸化得到产品,收率70%[3]。反应式为:
邬瑞斌等以不同方法制备了N-(2-氯-6-氟苯基)对甲苯胺,再经类似双氯芬酸钠的工艺合成了罗美昔布,酰化在甲苯中进行,收率86.4%,环合无溶剂操作,收率83.5%,水解在NaOH、乙醇、水中进行,收率75%[37]。李芬芬等用与文献[3]类似的方法合成了N-(2-氯-6-氟苯基)对甲苯胺,再经酰化、环合、水解制得罗美昔布,前两步无溶剂反应,收率分别为85.9%和83.1%,水解以PEG-2000催化,收率89.6%[38]。
1.3 卤代芳烃1步引入2个及以上碳原子单元反应
1.3.1 卤代芳烃与卤乙酸酯或卤乙酰胺的直接偶联反应法
卤代芳烃经金属有机试剂与卤乙酸衍生物直接偶联、水解是合成芳乙酸最有发展前景的方法之一,目前已报道的有格氏试剂偶联、有机锌试剂偶联和有机硼酸偶联法,一般使用镍或钯催化。良好的反应活性和选择性,价廉易得的催化剂和辅助配体,温和的反应条件,广泛的底物适用性是该合成法的关键。
张显飞等报道2,4,5-三氟溴苯与镁屑在四氢呋喃(THF)中反应制得2,4,5-三氟苯基溴化镁,再在氯化亚钴、四甲基乙二胺(TMEDA)催化下与溴乙酸乙酯偶联得到2,4,5-三氟苯乙酸乙酯,最后经稀NaOH水解、酸化得到2,4,5-三氟苯乙酸,总收率60%[39]。反应式为:
该方法存在原料价格较高,反应条件要求较为苛刻,催化剂氯化亚钴需要定量并且价格较高等缺点。
Bentz等报道,溴代芳烃或溴代杂环芳烃与溴乙酸酯或溴乙酰胺的有机锌(Reformatsky)试剂在四(三苯膦)合钯催化下和微波辅助下直接偶联得到芳乙酸酯或芳乙酰胺,水解可得芳乙酸。其中9例叔丁酯收率18%~98%,9例二苄基酰胺收率21%~84%,包括对位给电子或吸电子基团取代苯环及吡啶环[40]。
Liu等报道,苯硼酸与溴乙酸酯在摩尔分数5%的Ni(PPh3)4、4份K3PO4和甲苯中发生偶联生成芳乙酸酯,甲酯、乙酯和叔丁酯的收率分别为80%、93%和92%,水解可得芳乙酸[41]。
Peng等报道,芳硼酸与溴乙酸乙酯在钯催化和胺基膦配体作用下的交叉偶联反应中,水的存在极大地减少了芳硼酸的自身偶联,得到良好收率的芳乙酸乙酯。通过对溶剂、配体、钯催化剂和碱等因素的研究,得到THF、水为溶剂,二异丙基胺基二苯基膦(Ph2P-N(i-Pr)2)为配体,三(二亚苄基丙酮)二钯(Pd2(dba)3)为催化剂,碳酸钾为碱的效果最佳。在该反应条件下,13例芳乙酸乙酯的收率84%至98%,位阻较大的2,6-二甲基苯硼酸仍能获得60%收率的产物。水解可得芳乙酸[42]。反应式为(Ar为芳基):
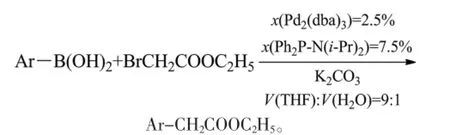

Zimmermann等报道,大立体位阻的苯硼酸与溴乙酸酯在摩尔分数分别为0.3%的Pd(dba)2和0.9%
三(邻甲苯基)膦催化下,KF为碱,苄基三乙基氯化铵为相转移催化剂作用下,发生偶联得到芳乙酸酯,19例收率33%至82%,水解得芳乙酸[43]。
1.3.2 卤代芳烃与草酸酯的格氏加成、还原反应法
卤代芳烃经与草酸酯的格氏加成及酮羰基还原、水解制得芳乙酸,是一种较好的合成方法,其中格氏试剂的制备和加成,酮羰基的还原和酯的水解均一锅完成,与1.1.2节法相同,而且草酸酯价廉易得。
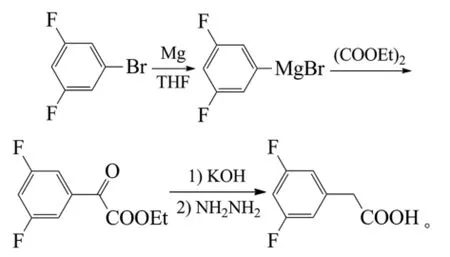
尹建湘等以3,5-二氟溴苯为原料,与镁粉制成格氏试剂,再与草酸二乙酯加成得到3,5-二氟苯乙酮酸乙酯,收率84%。KOH水解后,再经水合肼还原得3,5-二氟苯乙酸,收率81%[44]。反应式为:
方永勤等以4-三氟甲基苯胺为原料,经重氮化溴化得4-三氟甲基溴苯,与镁反应制成格氏试剂,再与草酸二乙酯加成制得4-三氟甲基苯乙酮酸乙酯,收率86.4%。最后在水合肼、KOH作用下制得4-三氟甲基苯乙酸,收率84.8%。较佳的合成工艺条件为:Mg与4-三氟甲基溴苯与草酸二乙酯摩尔配比为1.2:1:1.5、反应温度50~60℃、4-三氟甲基苯乙酮酸乙酯与水合肼摩尔配比为1:1.2[45]。
1.3.3 卤代芳烃与烯丙基溴的格氏偶联、断裂氧化反应法
卤代芳烃经格氏试剂与烯丙基溴偶联,再经C=C双键断裂氧化,得到芳乙酸,但氧化剂及催化剂价格太高,工业化价值有限。
Ikemoto等报道,2,4,5-三氟溴苯与异丙基氯化镁在THF中发生交换生成2,4,5-三氟苯基溴化镁,再与烯丙基溴偶联得到2,4,5-三氟烯丙基苯,最后在乙腈、水中经三氯化钌催化的高碘酸钠氧化制得到2,4,5-三氟苯乙酸[46]:

1.3.4 卤代芳烃与丙二酸酯和氰乙酸酯的亲核取代反应法
卤代芳烃,特别是邻/对位有强吸电子基团活化的卤代芳烃,与丙二酸酯或氰乙酸酯在适当的碱存在下发生卤素的亲核取代,再经水解脱羧制备芳乙酸,是一种应用较为广泛的合成方法。
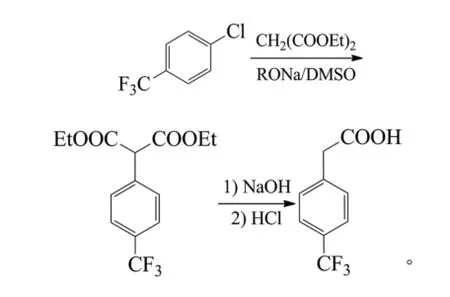
邱贵生等报道,对氯三氟甲苯和丙二酸二乙酯在叔丁醇钠催化下缩合、NaOH水解、浓硫酸酸化脱羧制得含氟中间体对三氟甲基苯乙酸,总收率75.4%[47]。反应式为:
葛裕华等报道,4-取代-2-硝基卤代苯与氰乙酸酯缩合、水解、脱羧等得到4-取代-2-硝基苯乙酸,其中卤素可以是氟、氯或溴,4位的取代基为氟、氯、溴、氰基、硝基、酯基和三氟甲基。如2,5-二溴硝基苯与氰乙酸乙酯在碳酸钠、丙酮和催化量碘化钾存在下反应,浓盐酸水解得到2-硝基-5-溴苯乙酸;2,5-二氟硝基苯与氰乙酸甲酯在碳酸钾、二甲基亚砜(DMSO)和催化量KI存在下反应,NaOH水解得到2-硝基-5-氟苯乙酸[48]。
徐大国等以2,5-二氯硝基苯为原料,经与丙二酸二乙酯缩合和选择性脱除一个酯基得到2-硝基-5-氯苯乙酸乙酯。该中间体再经硝基还原、环合、傅克酰化基酮基还原,得到新型抗精神病药齐拉西酮中间体。其中与丙二酸酯的缩合在NaH/DMF中进行,收率97.5%,选择性脱除单酯在LiCl/DMF中进行,收率85.0%[49]。
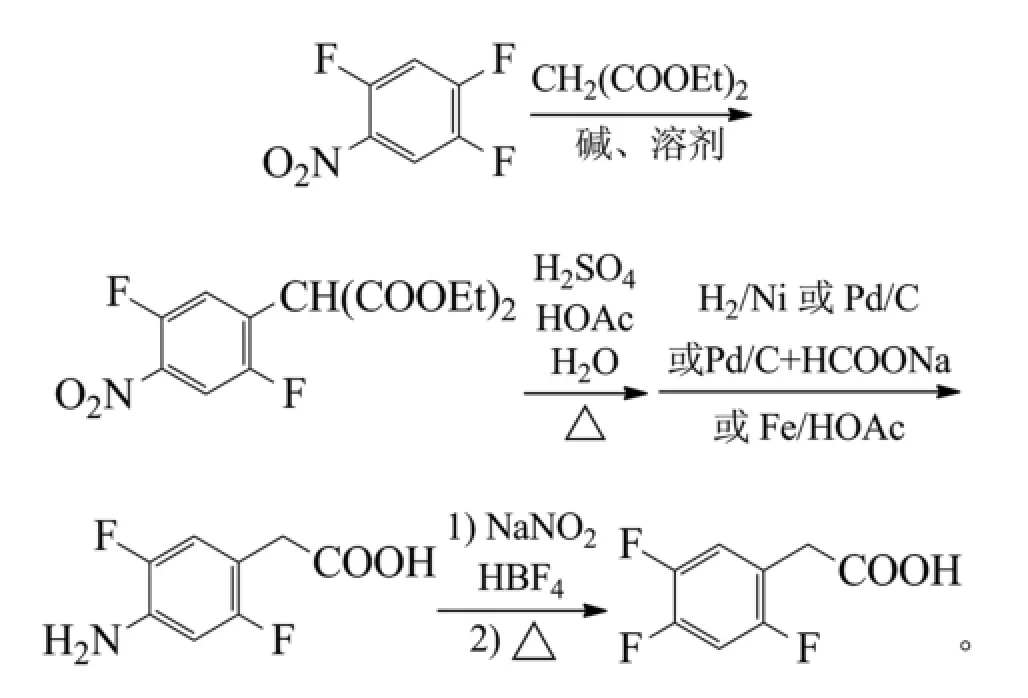
郑土才等以2,4,5-三氟硝基苯为原料,与丙二酸二乙酯在NaOH/DMF、KOH/N,N-二甲基乙酰胺、甲醇钠/N-甲基吡咯烷酮(NMP)等中缩合得到2,5-二氟-4-硝基苯基丙二酸二乙酯,再于硫酸、醋酸、水中回流得到2,5-二氟-4-硝基苯乙酸,2步收率约85%。2,5-二氟-4-硝基苯乙酸在5%的Pd/C、乙醇中常压加氢、雷尼镍/甲醇中加压氢化、5%的Pd/C、异丙醇中以甲酸钠为供氢体转移氢化、或铁粉、醋酸水溶液还原制得2,5-二氟-4-氨基苯乙酸,收率93%~
98%。最后经质量分数40%氟硼酸、亚硝酸钠重氮化、氟化制得2,4,5-三氟苯乙酸,收率58%~73%[50]。反应式为:
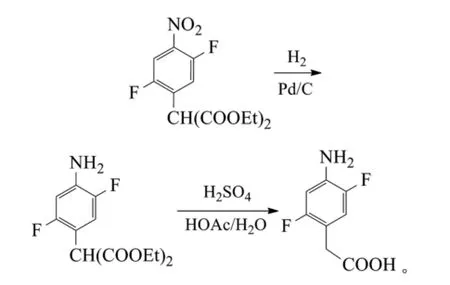
如前所得2,5-二氟-4-硝基苯基丙二酸二乙酯也可先经5%的Pd/C、乙醇中常压加氢、硫酸-醋酸-水中回流得到2,5-二氟-4-氨基苯乙酸,3步总收率88%。反应式为:
鲍樟水等报道1,2,4,5-四氟苯与氰乙酸乙酯经亲核取代、水解、脱羧等反应制得2,4,5-三氟苯乙酸,收率分别为70%和75%[51]。反应式为:
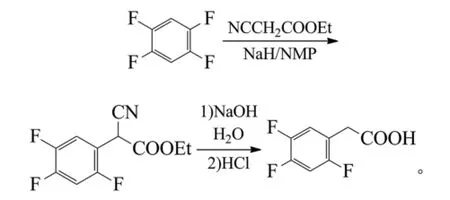
该方法所用原料四氟苯价格较高,氢化钠和NMP也较贵,氢化钠的使用存在安全隐患。
张亚梅等以异香兰醛为原料,在Br2、CHCl3作用下制得2-溴-3-羟基-4-甲氧基苯甲醛,收率93.2%。在乙酸酐、三乙胺作用下制得2-溴-3-甲酰基-6-甲氧基苯乙酯,收率96.3%。在对甲基苯磺酸、乙二醇作用下反应得到缩醛,收率97.7%。溴代苯和丙二酸二乙酯在乙醇钠为碱,六甲基磷酰三胺为溶剂和CuBr催化下,在氮气的保护下制得取代苯丙二酸二乙酯,收率39.1%。在浓盐酸、乙醇作用下进行反应制得1-羟基-2-甲氧基-6-甲酰基苯乙酸,收率95.6%[52]。
Zeevaart等报道,乙酰乙酸乙酯与碘代芳烃或缺电子的溴代芳烃在铜(I)催化下偶联直接合成芳乙酸乙酯,水解即得芳乙酸。反应不需额外配体,DMSO是优选溶剂,但1,4-二氧六环加乙二胺作配体也能得到较好的收率[53]。
Armstrong等报道,2,4,5-三氟溴苯与丙二酸二乙酯在叔丁醇钠、氯化亚铜、二氧六环或乙二醇二甲醚作用下缩合得2,4,5-三氟苯基丙二酸二乙酯,再经NaOH水解、盐酸酸化、加热脱羧制得2,4,5-三氟苯乙酸[54]:

该方法原料价格较高,溴的反应活性偏低,需要大量氯化亚铜催化,叔丁醇钠价格较高,如能进一步改进工艺,仍具有一定的工业化价值。
Latli等和Busacca等报道,氰基碳14标记的氰乙酸异丙酯与溴代芳杂环化合物在醋酸钯、三苯基膦、氢化钠、甲苯中反应,水解得到碳14标记的芳乙酸,总收率达96%[55-56]。反应式为:

Song等报道一系列溴代芳烃及缺电子氯代芳烃在Pd(dba)2、P(t-Bu)3、HBF4、K2CO3、KHCO3存在下与丙二酸二乙酯发生缩合和脱烷氧羰基化反应,得到芳乙酸乙酯,15例收率60%~95%,水解即可得芳乙酸。反应的催化剂易得,碱性温和,官能团相容性好,具有较好的应用前景[57]。
[1]吕早生,张琳涵,余腾飞,等.非甾体抗炎药联苯乙酸的合成综述[J].广州化工,2011,39(20):17-19.
[2]秦丙昌,陈静,廖新成,等.双氯芬酸钠合成工艺研究[J].应用化工,2008,37(3):275-278,297.
[3]张爱华,朱天卫,郭敏.罗美昔布的合成工艺改进[J].中国医药工业杂志,2006,37(4):221-222.
[4]张宗俭,李斌.世界农药大全:植物生长调节剂卷[M].北京:化学工业出版社,2011:204-206,166-168.
[5]杨柳阳,崔冬梅.苯乙酸类化合物的制备技术研究进展[J].浙江化工,2012,43(3):7-9.
[6]刘华平,曹自逸.苯乙酸合成研究进展[J].山东化工,2011, 40(5):25-27.
[7]束影.对羟基苯乙酸的合成及应用[J].广东化工,2010,37 (4):108-109.
[8]张鹏翔,张月成,李志陵,等.一种2,4,5-三氟苯乙酸的制备方法:中国,102584565[P].2012-07-18.
[9]邓绍平,陈洁.对三氟甲基苯乙酸的合成研究进展[J].山东化工,2012,41(4):47-49,52.
[10]彭勇,郑学忠,赵亚楠.瑞格列奈及其中间体合成的研究进展[J].河北师范大学学报:自然科学版,2010,34(1):89-92.
[11]李长波,赵国峥,张洪林.2-噻吩乙酸的合成技术进展及其应用[J].化学与生物工程,2006,23(7):4-6.
[12]韩会娟,李伟,何新蕾.利塞膦酸钠的合成[J].中国医药工业杂志,2008,39(4):241-243.
[13]刘长令.世界农药大全:杀虫剂卷[M].北京:化学工业出版社,2012:314-325.
[14]钟建新,何人报,王莺妹,等.2,3-二氟苯乙酸合成的制备方法:中国,101486638[P].2009-07-22.
[15]Brodney M A,Auperin D D,Becker S L,et al.Design, synthesis,and in vivo characterization of a novel species of tetralin amino imidazoles as-secretase inhibitors:discovery of PF-3084014[J].Bioorg Med Chem Lett,2011,21:2637-2640.
[16]Olguin J,Brooker S.Synthesis of 3-and 5-formyl-4-phenyl-1H-pyrazoles:promising head units for the generation of asymmetric imine ligands and mixed metal polynuclear complexes[J].New J Chem,2011,35:1242-1253.
[17]Alliot J,Gravel E,Pillon F,et al.Enantioselective synthesis of levo-milnacipran[J].Chem Commun,2012,48:8111-8113.
[18]Bury P S,Christiansen L B,Jacobsen P,et al.Synthesis and pharmacological evaluation of novel cis-3,4-diarylhydroxychromanes as high affinity partial agonists for the estrogen receptor[J].Bioorg Med Chem Lett,2002,10:125-145.
[19]Srinivasan B R,Shetgaonkar S Y,Nath Ghosh N.Synthesis and characterization of calcium(II)coordination polymers based on phenylacetic acid[J].J Coord Chem,2011,64(7): 1113-1124.
[21]Xu Y,McLaughlin M,Chen C,et al.A general method for the synthesis of 3,5-diarylcyclopentenones via Friedel-Crafts acylation of vinyl chlorides[J].J Org Chem,2009,74: 5100-5103.
[22]Basso A,Banfi L,Galatini A,et al.Isocyanides and arylacetic acids:synthesis and reactivity of 3-aryl-2-acyloxyacrylamides,an example of serendipity-oriented synthesis[J].Org Lett,2009,11(18):4068-4071.
[2]马冰洁,唐洪波.α-萘乙酸合成工艺研究[J].农药,2004,43 (9):412-413,416.
[23]柴多里,葛业峰,王国栋.植物生长调节剂α-萘乙酸的合成工艺研究[J].安徽化工,2007,33(1):27-28.
[24]陈刚,姚国新,朱锦桃.对甲氧基苯乙酸的合成新工艺研究[J].浙江理工大学学报,2011,28(1):151-154.
[25]姚国新,陈刚,卢勇.3,4-二甲氧基苯乙酸的合成新工艺[J].中国医药工业杂志,2010,41(10):730-731.
[26]崔庆荣.2-噻吩乙酸的合成[J].信阳农业高等专科学校学报,2008,18(2):136-137.
[27]向纪明,陈久存,李宝林.芳基乙酮酸乙酯的合成新方法[J].有机化学,2009,29(3):392-395.
[28]Ianni A,Waldvogel S.R.Reliable and versatile synthesis of 2-aryl-substituted cinnamic acid esters[J].Synthesis, 2006,(13):2103-2112.
[29]乔喜龙,田慧,李雯.乙醛酸合成对羟基苯乙酸的研究[J].河北工业科技,2005,22(5):264-266.
[30]李胜辉,夏菁,张立龙.对羟基苯乙酸的合成[J].河北大学学报:自然科学版,2012,32(3):265-268.
[31]张友恭,孙丽娟,王效峰.对羟基苯乙酸的合成[J].化学世界,2007(6):360-361,348.
[32]刘泽玲.2,4,5-三氟苯乙酸的合成研究[J].河北工业科技, 2011,28(4):244-246.
[33]Falchi A,Stivanello M,Serafini S.Process for the preparation of fluoro-phenylacetic acids and derivatives thereof:WO, 2008078350[P].2008-07-03.
[34]林艳艳.西他列汀中间体的合成研究[D].石家庄:河北科技大学,2010.
[35]李桂英,李亚巍,李英君.2-[(2,6-二氯苯基)氨基]苯乙酸钾的合成[J].长春工业大学学报:自然科学版,2007,28(4): 461-463.
[36]魏文珑,温艳珍,杜翠红,等.双氯灭痛合成新工艺的研究[J].太原理工大学学报,2004,35(6):710-713,716.
[37]邬瑞斌,陆涛.罗美昔布的合成研究[J].药学进展,2007,31 (12):573-575.
[38]李芬芬,王德才,江建.氯美昔布的合成研究[J].化工时刊, 2006,20(3):46-47,50.
[39]张显飞,李功勇,张国鑫,等.三氟苯乙酸的制备方法:中国,101450895[P].2009-06-10.
[40]Bentz E,Moloney M G,Westaway S M.Palladiumcatalyzed α-arylation of esters and amides under microwave conditions[J].Tetrahedron Lett,2004,45:7395-7397.
[41]Liu C,He C,Shi W,et al.Ni-catalyzed mild arylation of α-halocarbonyl compounds with arylboronic acids[J].Org Lett,2007,9(26):5601-5604.
[42]Peng Z Y,Wang J P,Cheng J,et al.Water works;an efficient palladium-catalyzed cross-coupling reaction between boronic acidsand bromoacetate with aminophosphine ligand[J].Tetrahedron,2010,66:8238-8241.
[43]Zimmermann B,Dzik W I,Himmler T,et al.Palladiumcatalyzed cross-coupling of sterically demanding boronic acids withα-bromocarbonyl com pounds[J].J Org Chem, 2011,76:8107-8112.
[44]尹建湘,陈红飙,赵辉.3,5-二氟苯乙酸的制备[J].中国医药工业杂志,2007,38(4):264-265.
[45]方永勤,徐冬梅.4-三氟甲基苯乙酸的合成新方法[J].化学试剂,2010,32(4):367-368,371.
[46]Ikemoto N,Dreher S D.Process for the synthesis of trifluorophenylacetic acids:US,2004077901[P].2004-04-22.
[47]邱贵生,杨芝.对三甲基苯乙酸的合成研究[J].化工时刊, 2011,25(6):23-25.
[48]葛裕华,王赟.2-硝基-4-取代苯乙酸的合成方法:中国, 101805265[P].2010-08-18.
[49]徐大国,蒋成君.5-(2-氯乙基)-6-氯-1,3-二氢吲哚酮的合成[J].浙江化工,2013,44(7),14-16.
[50]郑土才,吾国强,吕延文.一种2,4,5-三氟苯乙酸的制备方法:中国,103012111[P].2012-09-12.
[51]鲍樟水,冯启,夏旭建,等.2,4,5-三氟苯乙酸的合成[J].浙江化工,2014,45(1):4-6.
[52]张亚梅,郭永恩,王婷婷.丹酚酸B重要中间体的合成和表征[J].安徽农业科学,2009,37(34):16749-16750.
[53]Zeevaart J G,Parkinson C J,de Koning C B.Copper(I) iodide-catalyzed arylation of acetoacetate to yield 2-arylaceticacid esters[J].Tetrahedron Lett,2007,48:3289-3293.
[54]Armstrong J D,Dreher S D,Ikemoto N.Process for the synthesis of trifluoro-phenylacetic acids:US,2004/068141 [P].2004-04-08.
[55]Latli B,Hrapchak M,Busacca C A,et al.Synthesis of14C and13C6-labeled potent HIV non-nucleoside reverse transcriptase inhibitor[J].J Label Compd Radio-pharm, 2009,52:84-90.
[56]Busacca C A,Cerreta M,Dong Y,et al.Development of a pilot-plant process for a nevirapine analogue HIV NNRT inhibitor[J].Org Proc Res Dev,2008,12:603-613.
[57]Song B,Rudolphi F,Himmler T,et al.Practical synthesis of 2-arylacetic acid esters via Palladium-catalyzed dealkoxycarbonylative coupling of malonates with aryl halides[J]. Adv Synth Catal,2011,353:1565-1574.
(待续)(吴红富)
运用超微粉体技术抢占绿色环保装备制高点浙江丰利通过省级企业技术中心复评
日前,国家高新技术企业浙江丰利粉碎设备有限公司企业技术中心,经浙江省经信委、省财政厅、省国税局、省地税局和杭州海关等5部门联合审定,得分高于75分,符合浙江省省级企业技术中心创新能力建设项目申报得分条件,顺利通过复评。据悉,省级企业技术中心评定不仅在企业销售规模等经济指标上设有较高门槛,更注重企业的创新能力、创新示范作用等综合水平,同时要求企业在行业中具有显著的规模优势和竞争优势。省有关部门予以省级认定,并给予相应的政策扶持。
近年来,随着科学技术的不断进步,材料技术得到了飞速发展,复合材料作为一种新型材料已经逐渐成为21世纪的主导材料之一。但其废弃材料也已然成为亟待解决的社会问题。为此,浙江丰利技术中心坚持绿色为先战略,专门成立废弃生物质资源化及装备工程技术中心,运用先进粉体技术进行破解,取得了可喜成果:承担的浙江省重大科技专项 “废电器及电子材料综合利用技术及其成套设备”实现了废电器及电子材料综合利用技术的重大突破;前不久,工业和信息化部、科技部和环境保护部三部委联合发布了《国家鼓励发展的重大环保技术装备目录(2014年版)》的通告。浙江丰利开发的“废塑料复合材料回收处理成套装备”入选该《目录》推广类条目下“五、资源综合利用”项目,适用废塑料综合利用范围。
目前,浙江丰利名符其实成为我国粉体设备及绿色环保装备供应商,闻名海内外的成套超微粉体设备生产基地。产品畅销全国,远销欧美、东南亚等国家和地区。
TQ245.1
A
10.3969/j.issn.1006-6829.2015.01.012
衢州市科技计划项目(2013Y001),浙江省大学生科技创新项目(2014R427001)*
。电子邮件:tczheng 2004@aliyun.com
2014-09-23
猜你喜欢
食管疾病(2022年1期)2022-11-26
浙江化工(2022年8期)2022-09-05
酿酒科技(2022年8期)2022-08-20
有机氟工业(2020年2期)2020-07-04
中国卫生标准管理(2015年14期)2016-01-15
烟草科技(2015年8期)2015-12-20
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
医学研究杂志(2015年9期)2015-07-01
云南中医学院学报(2014年2期)2014-11-07
