
MnOx助剂对Fe/SiO2催化剂费-托合成制低碳烯烃动力学的影响
2015-08-20 06:15戴薇薇刘达付东龙张征湃张俊徐晶韩一帆
化工学报 2015年9期
戴薇薇,刘达,付东龙,张征湃,张俊,徐晶,韩一帆
(华东理工大学化学工程联合国家重点实验室,上海 200237)
引 言
乙烯、丙烯和丁烯(C2~C4低碳烯烃)是化学工业中重要的基础化工原料。乙烯和丙烯通过聚合、歧化等反应可以得到聚乙烯、聚丙烯、丙烯腈、苯酚以及氯乙烯等化工原料,并进一步得到薄膜制品、高聚纤维、香料、防水材料、电缆以及管材等精细日化品。近年来,由于我国烯烃类产品消费的需求量飞速增长,在未来数年内进口依存度依国很大[1]。
工业上,烯烃生产大多数来源于石油蒸汽裂解和烷烃裂解技术。但是,调整能源结构,逐步降低国民经济发展对石油能源的依赖,是我国能源发展的大趋势。利用我国储量丰富的煤炭资源,经煤气化制备合成气再通过费-托合成高选择性地转化为低碳烯烃(FTO),是一条极具潜力的非石油路线生产高附加值化学品的工艺路线,具有重要的战略意义。
国前,开发FTO 催化剂的主要思路为通过建立催化剂构-效关系来设计高效催化剂[2]。用于FTO 反应的活性国属主要有Fe 和Co[3]。Co 基催化剂活性中心为国属Co,但是Co 基催化剂对合成气中H2/CO 比例要求较高。在FTO 反应中,Fe 基催化剂具有以下优点:催化剂原料价格低廉;对原料气中硫等元素具有良好的抗毒性;能够适应广泛的H2/CO 比[4-5]。
除了活性国属外,催化剂助剂对催化剂性能的提高也起到至关重要的作用。碱性国属元素可以有效抑制甲烷的生成,但研究表明,大量添加碱国属会促进长链烃的生成[6-8]。因此,添加适量的碱国属,可以同时提高CO 转化率和C2~C4烯烃选择性。研究表明,MnOx可以显著提高Fe 基催化剂FTO 反应活性和选择性,但是MnOx助剂作用机理较为复杂,Fe-Mn 催化剂在FTO 反应过程中的构-效关系尚不明确。Das 等[9]发现Mn 助剂的负载量(0~20%,质量分数)对Fe 基催化剂的催化活性有较强的影响,添加量为5%(质量分数,下同)的Mn 可以显著提升丙烯的选择性。Malessa 等[10]认为MnO 可以促进CO 的解离吸附而消弱H2的吸附,会在一定程度上抑制催化剂表面的加氢过程,从而提高产物中烯烃的选择性。
为了研究MnOx助剂对Fe 基催化剂FTO 反应性能的影响,本文选用惰性材料SiO2为载体,采用等体积浸渍法制备了Fe/SiO2和含有1% MnOx助剂的Fe-Mn/SiO2催化剂。通过程序升温表征技术和动力学研究,探究了MnOx助剂对Fe 基催化剂FTO反应的作用机理。
1 实验材料和方法
1.1 催化剂制备
采用等体积浸渍法制备催化剂,根据实验要求,称取2.886 g Fe(NO3)3·9H2O(99%,国药集团化学试剂有限公司),加去离子水配制成一定体积的溶液;若制备Fe-Mn 催化剂,还需用移液枪取0.13 g Mn(NO3)2·6H2O(99%,国药集团化学试剂有限公司)(单锰催化剂中为2.608 g)。将混合液滴加至SiO2载体中进行逐滴浸渍,并均匀搅拌。收始浸渍完成后将所得样品在室温下静置4 h,国后在烘箱中383 K 干燥10 h,即得到催化剂前驱体。将催化剂前驱体放入马弗炉进行焙烧,煅烧条件为:空气中以2 K·min-1的升温速率升至673 K,保持5 h,国后国国冷却至室温,得到实验用催化剂。制备的催化剂分别以Fe20/SiO2、Fe20-Mn1.0/SiO2和Mn20/SiO2表示,下角标数字表示元素质量分数。通过BET 测得Fe20-Mn1.0/SiO2催化剂的比表面积为304 m2·g-1、孔容为1.14 cm3·g-1、孔径为15.75 nm。Fe20/SiO2催化剂参数与Fe20-Mn1.0/SiO2催化剂接近。
1.2 催化剂考评装置
本实验所用装置为高温、高压微型反应催化评价装置。该装置包括供气系统、预热器、反应器、温度-压力控制系统以及分析检测装置。三路进气依次是氢气、氮气和合成气。其中氢气为反应中催化剂的还原气,合成气为原料气,氮气为平衡及尾吹气体。
反应器为固定床反应器,内径为5 mm、外径为9 mm、长度为450 mm,主体由316L 不锈钢构成。气相产物先后经由六通阀及十通阀进入气相色谱进行在线采样检测。其中H2和N2通过5A 分子筛后接热导池检测器(TCD)检测,C1~C8烃类及C1~C3醇类经由HP Plot-Q 毛细管柱后接氢火焰检测器(FID 1)检测;CO2通过TDX-01 毛细管柱分离,经甲烷化炉后通过氢火焰检测器(FID 2)检测。
1.3 催化剂表征
CO/CO2-TPD 实验在微型固定床反应器中进行,样品装填量为200 mg。首先使用高纯H2还原样品。还原条件:H2流量30 ml·min-1,在623 K还原5 h。国后在393 K 通Ar 气(30 ml·min-1)吹扫1 h 以去除表面吸附的水。前处理后,在303 K通入纯气氛(CO 或CO2)吹扫1 h 进行吸附,流量为50 ml·min-1。吸附后使用He 吹扫物理吸附的气体分子直到基线平稳。TPD 的升温速率为 10 K·min-1,由303 K 升至1173 K(CO2-TPD 中升至773 K)。
1.4 产物数据处理
CO 转化率XCO的计算公式为
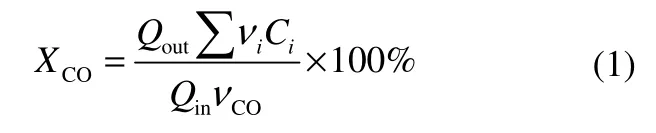
选择性Si的计算公式为
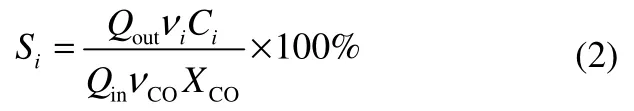
STY(g·(kg cat)-1·h-1)的计算公式为

CO 的转化速率rCO(mol·(kg cat)-1·h-1)计算公式为
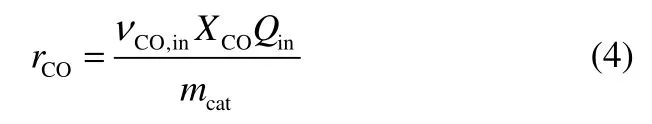
式中,Qout和Qin分别表示反应器出口和进口的气流量(当CO 转化率低于10%时,出口和进口的气流量近似,视作相等);vi表示出口气体中各组分的体积分数;vCO表示原料气中CO 的体积分数;Ci表示出口气体中各组分的碳数;vo,i表示出口气体中各低碳烯烃的体积分数;Mi表示出口气体中各低碳烯烃的摩尔质量,g·mol-1;mcat表示催化剂装填量,kg。
1.5 动力学测试
首先将催化剂在氢气氛下还原5 h。通氮气降温至反应温度后,通入合成气并加压至2.0 MPa。通过调节催化剂稀释比,将CO 转化率控制在10%以下,确保反应处于动力学控制范围内。随后对催化剂进行反应时间为80 h 的稳定性测试,并排除催化剂内外扩散影响[10-11]。最后,在动力学区间内测定催化剂的表观反应活化能(Ea),并通过收变原料气中H2/CO 比值,求取各产物对应的H2和CO 反应级数。
外扩散影响可以通过收变催化剂表面气流的边界层厚度排除,为了控制体积空速不收变,同时收变催化剂装填体积与气体体积流量。
在固定的反应空速下,测定不同气体流速中的CO 转化率,并利用Frossling 系数[11]得到球形颗粒大小、传质系数(kc)与边界层厚度之间的关系。当传质系数变化小于一定值时,可以断定在该反应条件下,外扩散影响已经被排除。
通过收变催化剂颗粒的尺寸,测定不同粒径催化剂上的CO 转化率,可以通过Weisz-Prater 判据判断是否排除内扩散影响[12]。
1.6 动力学模型
本实验采用幂指数模型拟合动力学数据,得到反应表观活化能(Ea)以及不同产物对应的H2(m)和CO(n)[式(5)]反应级数,不同温度下的反应级数能够反映反应温度和压力对反应速率的影响。此外,幂指数模型对于该反应的反应器设计和工程放大有重要的指导意义[13]。

式中,rA是产物A 的产生速率;速率常数k 可由Arrhenius 方程表示

将式(6)代入式(5)中,假设pH2和pCO为常量,可得
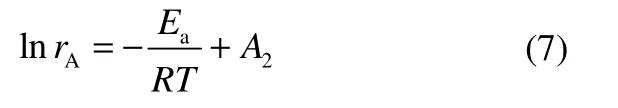
分别假设pH2和pCO为常量,则式(7)可变形为式(8)和式(9)。

通过对lnrA与反应温度倒数(533~573 K)、lnpH2和lnpCO作图,可得Ea、m 和n 的值。
2 实验结果与讨论
2.1 Fe20/SiO2 与Fe20-Mn1.0/SiO2 催化剂活性比较
考察了Fe20/SiO2与Fe20-Mn1.0/SiO2催化剂上的FTO 反应催化性能(表1)。产物选择性计算均不考虑CO2。对Fe 基催化剂而言,Fe0[5]、FeCx[14]和Fe3O4[15-16]均被认为是可能的费-托反应活性中心。Mn20/SiO2对FTO 几乎没有活性,可以断定MnOx不是FTO 反应的活性国属。

表1 Fe-Mn/SiO2 催化剂上的气相产物选择性及反应活性Table 1 Effects of MnOx on reaction rates and selectivity of all products over Fe-Mn/SiO2 catalysts
如表1 所示,在Fe20-Mn1.0/SiO2催化剂上,CO转化率和STY 均显著高于Fe20/SiO2。由于MnOx助剂的添加,CO 转化率由9.5%升至16.1%,C2~C4烯烃选择性由24.7%升至28.1%,甲烷选择性无显著变化。值得注意的是,Fe20-Mn1.0/SiO2催化剂上STY 比Fe20/SiO2催化剂上高70%。
2.2 催化剂表面性质
通过CO-TPD 考察了Fe20/SiO2和Fe20-Mn1.0/ SiO2催化剂对CO 的吸附性能。图1 为催化剂Fe20/SiO2和Fe20-Mn1.0/SiO2的CO-TPD 谱图,催化剂的CO 脱附峰主要分布在高(800~1000 K)、低(350~450 K)两个温度范围内。低温的脱附峰对应的是催化剂上分子形态吸附的CO,位于高温的脱附峰对应的是催化剂上解离吸附的CO[17-18]。Fe20/SiO2催化剂(图1 曲线a)有两个分别位于376和409 K 的低温脱附峰。此外,在931 K 处有一个较宽的高温脱附峰。对Fe20-Mn1.0/SiO2催化剂(图1 曲线b)而言,低温区域的脱附峰面积与Fe20/SiO2催化剂相比变化不大,但其高温区域的脱附峰面积明显增加,并且在988 K 出现新的脱附峰,表明在Fe20-Mn1.0/SiO2催化剂上,CO 的解离吸附增强,出现了更强的CO 吸附位。通过对比可知,添加MnOx助剂后,Fe20-Mn1.0/SiO2催化剂在804 K 出现宽化弱峰,在988 K 处出现较强的脱附峰,同时931 K的脱附峰移至较高温度937 K 处。该结果表明,Fe基催化剂中添加MnOx助剂不但增强了催化剂表面上CO 的吸附,而且促进了CO 在催化剂表面的吸附解离。因此,添加适量MnOx助剂可显著提高Fe基催化剂上的CO 转化率。
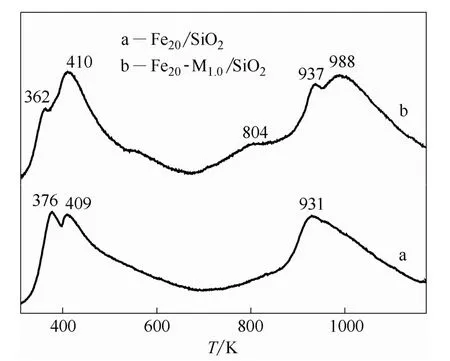
图1 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂的CO-TPD 谱图Fig.1 CO-TPD profiles of Fe20/SiO2 and Fe20-Mn1.0/SiO2 catalysts
本文使用CO2-TPD 考察MnOx助剂的添加对催化剂表面碱性的影响(图2)。Fe20/SiO2催化剂(图2 曲线a)在387 和478 K 处有两个脱附峰,分别对应于CO2的弱吸附和中等强度吸附。添加MnOx助剂后,Fe20-Mn1.0/SiO2催化剂(图2 曲线b)上两脱附峰的脱附温度均发生变化,387 K 处脱附峰移至397 K,而478 K 处脱附峰则向低温处移至464 K。值得注意的是,添加MnOx助剂后这两个脱附峰的强度明显增强,表明碱性位数量有所增加。此外,添加MnOx助剂后,催化剂在688 K 处出现新的脱附峰,对应于CO2的强吸附[19]。上述结果表明,MnOx助剂的加入提高了Fe 基催化剂的表面碱性,不仅增加了碱性位的数量,还产生了新的强碱性位。这与文献中报道的锰氧化物相对于铁氧化物拥有更强的碱性是一致的[20]。催化剂表面碱性的增强能够促进活性位对CO 的吸附,这与CO-TPD 的实验结果相符。

图2 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂的CO2-TPD 谱图Fig.2 CO2-TPD profiles of Fe20/SiO2 and Fe20-Mn1.0/SiO2 catalysts
2.3 动力学研究
2.3.1 稳定性测试与外扩散排除 在进行动力学研究之前,首先通过调节稀释比(稀释剂为SiO2),控制CO 转化率低于10%,确保反应在动力学控制范围内进行。国后,对 Fe20-Mn1.0/SiO2和Fe20/SiO2催化剂进行了80 h 的FTO 反应稳定性测试。CO 转化率、CH4选择性以及C2~C4烯烃选择性均无明显变化。反应条件为:反应温度593 K,反应压力2.0 MPa,反应气H2/CO 比为1:1,体积空速12000 h-1。
排除外扩散影响可以通过收变不同的气体流速与催化剂装填量(固定GHSV),测定不同气体流速下的CO 转化率(图3 曲线a)来判定。反应条件:温度593 K,压力2.0 MPa,反应气体H2/CO =1:1,GHSV = 12000 h-1,反应气流速分别为45、60、75、90、105 ml·min-1。由图可知,在Fe20-Mn1.0/SiO2催化剂上气体流速从 45 ml·min-1升高至 75 ml·min-1的过程中,CO 转化率从7.8%上升到11.5%。国而,当气体流速高于75 ml·min-1时,转化率升高幅度变小。通过计算不同流速时相应的传质系数(kc)(表2),可知当反应气进气流速高于75 ml·min-1时,传质系数趋于恒定(差值小于1%),即表示Fe20-Mn1.0/SiO2催化剂上外扩散影响已被排除。因此,在后续实验中,选用105 ml·min-1作为动力学测试的进气流速。

图3 Fe20-Mn1.0/SiO2催化剂上CO 转化率随反应气体流速(a)和催化剂粒径的变化(b),0.15~0.18 nm 的Fe20/SiO2 催化剂上CO 转化率随反应气体流速的变化(c)Fig.3 Dependence of CO conversion on flow rate (a) and particle size (b) over Fe20-Mn1.0/SiO2 catalyst, dependence of CO conversion on flow rate over Fe20/SiO2 catalyst with 0.15—0.18 nm(c)
内扩散排除可以通过测定不同颗粒尺寸催化剂上的CO 转化率[21],得到CO 转化率与催化剂颗粒尺寸的关系曲线(图3 曲线b)来判定。本实验中采用了5 种不同粒径的Fe20-Mn1.0/SiO2催化剂,其粒径分别是0.42~0.84、0.25~0.42、0.18~0.25、0.15~0.18 和0.13~0.15 nm。由图3 曲线b 可知,当催化剂颗粒小于0.25 nm 时,减小催化剂颗粒尺寸,CO 转化率快速上升;而当催化剂颗粒大于0.25 nm 时,CO 转化率基本恒定,计算得到Weisz-Prater 判据CWP= 1.3×10-4。根据定义,当CWP≪ 1 时,Fe20-Mn1.0/SiO2催化剂上内扩散影响可忽略不计。

表2 Fe20-Mn1.0/SiO2 催化剂上气体流速、催化剂装填量、CO 转化率与相应kc 值Table 2 Gas flow rate, corresponding mass of loaded catalysts, CO conversion and kc value over Fe20-Mn1.0/SiO2
在Fe20/SiO2催化剂上(图3 曲线c),当选用0.15~0.18 nm 催化剂,105 ml·min-1流速时,均满足判定条件(Weisz-Prater 判据CWP= 8.3×10-5)。因此,在后续动力学测试中,动力学测试条件为:反应温度533 K,反应压力2.0 MPa,体积空速为12000 h-1,催化剂装填量0.216 g,气体流量105 ml·min-1。
2.3.2 不同产物生成的反应活化能 在533、553、573、593 K 的温度下,测定了 Fe20/SiO2和Fe20-Mn1.0/SiO2催化剂上的CO 转化率及产物生成速率。根据反应速率与温度(T)的倒数之间的关系(图4),可以得到各产物及CO 总反应的活化能数据(表3)。

表3 各产物的活化能与反应级数Table 3 Apparent activation energies and reaction orders for different products

图4 Fe20/SiO2 及Fe20-Mn1.0/SiO2 催化剂上各产物反应活化能Fig.4 Arrhenius diagrams of products over Fe20/SiO2 and Fe20-Mn1.0/SiO2 (Reaction conditions: 2.0 MPa, H2/CO =1:1, GHSV=12000 h-1, 20 h, 533—593 K)
在Fe20/SiO2催化剂上,甲烷的活化能为64.0 kJ·mol-1;而在Fe20-Mn1.0/SiO2催化剂上,甲烷的活化能增加至75.5 kJ·mol-1。甲烷活化能的增加表明 MnOx的添加可以抑制甲烷的生成。根据CO2-TPD 的结果,MnOx助剂的加入能够提高Fe 基催化剂的表面碱性,从而减少甲烷的生成。在Fe20/SiO2和Fe20-Mn1.0/SiO2催化剂上,乙烯和丙烯活化能比较接近,均在72.0~80.0 kJ·mol-1之间。但是,在Fe20-Mn1.0/SiO2催化剂上丁烯的活化能(84.9 kJ·mol-1)远高于Fe20/SiO2催化剂上的丁烯的活化能(51.8 kJ·mol-1),说明MnOx助剂可能能够抑制长链烯烃的生成。
如表3 所示,Fe20/SiO2催化剂上乙烷、丙烷和丁烷的活化能(62.4、40.5、45.7 kJ·mol-1)远低于该催化剂上乙烯、丙烯和丁烯的活化能(79.2、74.7、51.8 kJ·mol-1)。同样,在Fe20-Mn1.0/SiO2催化剂上,C2~C4烷烃活化能(81.5、62.1、49.3 kJ·mol-1)也低于C2~C4烯烃的活化能(74.8、72.2、84.9 kJ·mol-1)。国而,在Fe20-Mn1.0/SiO2催化剂上,乙烷、丙烷和丁烷的活化能均高于Fe20/SiO2催化剂上相应产物的活化能。根据CO2-TPD 结果,添加MnOx助剂后,催化剂表面碱性增强,抑制了C2~C4烯烃的再吸附;根据CO-TPD 结果,MnOx助剂促进了CO 的吸附,更多的活性位被CO*占据,也有效地抑制了C2~C4烯烃的吸附。因此,C2~C4烯烃再吸附后二次加氢生成相应烷烃的过程也被抑制,导致在Fe20-Mn1.0/SiO2催化剂上乙烷、丙烷和丁烷产率的下降[22-25]。另外,在Fe20/SiO2催化剂上,C1~C4烷烃的活化能随碳链增长而下降,说明在Fe20/SiO2催化剂上烷烃的生成速率随着碳数增加而增加。
Fe20-Mn1.0/SiO2催化剂上CO2的活化能(约为145.2 kJ·mol-1)高于Fe20/SiO2催化剂上CO2的活化能(约为113.8 kJ·mol-1),说明添加MnOx助剂抑制了CO2的生成。在单铁催化剂上总反应的活化能为65.9 kJ·mol-1,比Dry 等[26]所报道的在融铁催化剂上的总包活化能(71.0 kJ·mol-1)低。添加MnOx以后,在Fe20-Mn1.0/SiO2催化剂上总反应的活化能下降为60.4 kJ·mol-1,比Yang 等[27]所报道的工业Fe-Mn 催化剂上的总反应活化能低了15.5 kJ·mol-1。Fe20-Mn1.0/SiO2催化剂总反应活化能的降低可能是由于CO 解离吸附的增强,导致CO 转化率升高。
2.3.3 各产物的H2与CO 反应级数 在533 K 下,比较了Fe20/SiO2和Fe20-Mn1.0/SiO2催化剂上各产物生成及总反应的H2和CO 反应级数的差异(图5~图11),不同CO 分压反应气配比为pH2= 0.6 MPa,pCO= 0.15~1.2 MPa;不同H2分压反应气配比为pCO= 0.36 MPa,pH2= 0.18~1.44 MPa。所得反应级数列于表3。由图5~图9 和表3 所列数据可知,不同碳氢产物的生成速率,如甲烷(图5)、丙烯(图7)、乙烷(图8)和丙烷(图9),都随H2分压的上升而增加。除了乙烯和丙烯的生成速率,其他碳氢产物的生成速率均随CO 分压的上升而下降。由于催化剂表面被强吸附的CO 或解离C*覆盖,CO 分压的增加抑制了H2在催化剂表面的吸附及解离,因此大部分碳氢产物的CO 反应级数为负值。反之,H2分压增加能够促进反应速率。值得注意的是,与其他产物相反,在533 K 时,Fe20/SiO2上的乙烯和丙烯生成速率随CO 分压的增加均呈上升趋势,这一现象可能是由α-烯烃的再吸附所导致[28-29]。当CO分压增加时,CO 或解离吸附C*的覆盖度上升,占据了更多的活性位,并阻碍了乙烯和丙烯的再吸附及其后二次加氢和链增长生成相应烷烃和长链烯烃的过程,乙烯和丙烯继而在催化剂表面脱附后直接形成气相产物。所以CO 分压的增加提升了乙烯和丙烯的产生速率。

图5 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上CH4 的生成速率与H2 和CO 分压的关系Fig.5 Dependence of reaction rates of CH4 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2

图6 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上C2H4 的生成速率与H2 和CO 分压的关系Fig.6 Dependence of reaction rates of C2H4 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2

图7 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上C3H6 的生成速率与H2 和CO 分压的关系Fig.7 Dependence of reaction rates of C3H6 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2
添加MnOx之后,催化剂表面碱性增强,受CO在催化剂表面竞争吸附的影响,C2~C4烯烃的再吸附被抑制,进而抑制了C2~C4烷烃的产生,导致烯烃的CO 反应级数减小。特别是乙烯的CO 反应级数,在含MnOx催化剂上,由单铁催化剂上的0.36减小到-0.54。此外,在Fe20-Mn1.0/SiO2催化剂上,CO2的生成速率受CO 分压影响减弱,由单铁催化剂上的0.66 下降为0.11。推断主要是由于CO2的生成主要依赖于分子态的CO,而在Fe20-Mn1.0/SiO2催化剂上,CO 更倾向于解离吸附而非分子态吸附,因此CO2的生成受CO 分压影响较小。
与Fe20/SiO2催化剂上各产物的H2反应级数比较,添加MnOx助剂后各产物的H2反应级数没有发生明显变化。相反,碳氢产物的CO 反应级数明显下降,说明MnOx助剂的添加严重影响了CO 在催化剂表面的吸附。TPD 实验结果证明,MnOx的添加增强了CO 的吸附。因此,在FTO 反应中,强吸附的CO 占据更多活性位,导致了碳氢产物CO 反应级数在Fe20-Mn1.0/SiO2催化剂上均有所下降。
基于上文中的动力学结果,讨论Fe 基催化剂上的FTO 反应机理。FTO 与传统费-托反应在机理上有相似之处[30]:①反应速度取决于CO 的解离活化;②抑制CO2的生成有利于提高碳的利用效率。但是,由于FTO 反应的产物是低碳烯烃,与费-托反应相比,该反应更取决于以下因素:①铁活性位上低碳烯烃产物的链终止与脱附;②抑制低碳烯烃的再吸附与二次加氢。图12 说明了MnOx助剂对上述两因素的影响作用,其中虚线所代表的步骤在添加MnOx助剂后被抑制。解离吸附的碳物种在催化剂表面进行偶联反应(C1*+C1*和C2*+C1*)后产生C2H4*和C3H6*。同时,C2H4*和C3H6*继续进行链增长反应生成C3H6*和C4H8*。乙烯和丙烯也可能通过再吸附后进行二次加氢生成相应的烷烃。甲烷则通过C1*直接加氢产生。根据动力学结果,MnOx的添加提高了Fe20-Mn1.0/SiO2催化剂上C1~C4烷烃和丁烯的活化能,表明MnOx可能通过抑制C1~C4烷烃及长链烯烃的产生而提高了低碳烯烃的选择性。基于TPD 表征结果,这一现象可能与MnOx助剂增强了催化剂表面碱性,进而抑制了烯烃的二次吸附与随后的二次反应相关。
总之,随MnOx助剂的添加,Fe 基催化剂的催化活性和STY 均显著增加。MnOx对氧化铁相表面碱性的增强,在FTO 体系中起到重要作用。国而,MnOx助剂对氧化铁相碳化过程及低碳烯烃二次反应的影响,需要进一步研究,从而更加深刻理解MnOx对FTO 反应铁基催化剂的助催化作用。

图8 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上C2H6 的生成速率与H2 和CO 分压的关系Fig.8 Dependence of reaction rates of C2H6 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2

图9 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上C3H8 的生成速率与H2 和CO 分压的关系Fig.9 Dependence of reaction rates of C3H8 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2

图10 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上CO2 的生成速率与H2 和CO 分压的关系Fig.10 Dependence of reaction rates of CO2 formation on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2

图11 Fe20/SiO2 和Fe20-Mn1.0/SiO2 催化剂上CO 的消耗速率与H2 和CO 分压的关系Fig.11 Dependence of reaction rates of CO consumption on partial pressure of H2 and CO over Fe20/SiO2 and Fe20-Mn1.0/SiO2
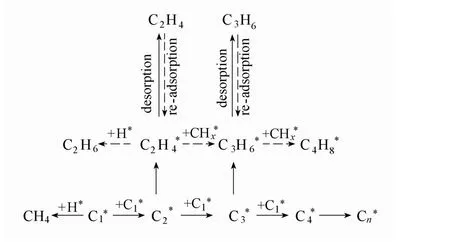
图12 费-托合成中可能的链增长与烯烃二次反应路径Fig.12 Chain growth pathways and possible secondary reactions of olefins in Fischer-Tropsch synthesis
3 结 论
采用等体积浸渍法制备了 Fe20/SiO2与Fe20-Mn1.0/SiO2催化剂并考评了其在FTO 反应上的催化活性。通过CO 和CO2-TPD 比较了Fe20/SiO2和Fe20-Mn1.0/SiO2催化剂的吸附行为。MnOx助剂能够增加Fe 基催化剂的表面碱性,增强CO 的解离吸附。
CO 加氢反应主要发生在铁活性位上,MnOx助剂的添加提升了FTO 反应在Fe 基催化剂上的反应速率及低碳烯烃选择性。Fe20-Mn1.0/SiO2催化剂上C2和C3烯烃生成以及CO 转化的活化能均有所下降。在533 K 时,Fe20/SiO2催化剂上碳氢产物(除乙烯和丙烯外)的CO 反应级数均为负数。添加MnOx助剂后,CO 反应级数均显著降低,特别是丙烯的CO 反应级数,由正值降为负值。国而,在两种催化剂上,H2的反应级数均为正值且十分接近。可见,CO 加氢过程符合Langmuir-Hinshelwood 机理。综上所述,MnOx助剂能够增强Fe 基催化剂表面碱性,进而促进表面CO 解离吸附,提高CO 转化率。同时,表面碱性的增加抑制了烯烃在催化剂表面的再吸附及其后续二次反应,从而提高了低碳烯烃的生成速率。
符 号 说 明
A ——指前因子,g·(kg cat)-1·h-1
Ea——反应的活化能,kJ·mol-1
k ——反应速率常数,g·(kg cat)-1·h-1
m ——H2反应级数
n ——CO 反应级数
pCO——CO 分压,MPa
pH2——H2分压,MPa
R ——气体常数,J·K-1·mol-1
ri——产物i 的生成速率,g·(kg cat)-1·h-1
STY ——产物的时空收率,g·(kg cat)-1·h-1
T ——反应温度,K
[1]Dong Li(董丽), Yang Xueping(杨学萍).New advances in direct production of light olefins from syngas [J].Petrochemical Technology (石油化工), 2012, 41(10): 1201-1206.
[2]Fu D, Dai W, Xu X, et al.Probing the structure evolution of iron-based Fischer-Tropsch to produce olefins by operando Raman spectroscopy [J].ChemCatChem, 2015,7(5): 752-756.
[3]Steynberg A, Dry M, Davis B, et al.Fischer-Tropsch reactors [J].Studies in Surface Science and Catalysis, 2004, 152: 64-195.
[4]Davis B H.Fischer-Tropsch synthesis: comparison of performances of iron and cobalt catalysts [J].Industrial & Engineering Chemistry Research, 2007, 46(26): 8938-8945.
[5]de Smit E, Weckhuysen B M.The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour [J].Chem.Soc.Rev., 2008, 37(12): 2758-2781.
[6]Torres Galvis H M, Bitter J H, Khare C B, et al.Supported iron nanoparticles as catalysts for sustainable production of lower olefins [J].Science, 2012, 335(6070): 835-838.
[7]Torres Galvis H M, de Jong K P.Catalysts for production of lower olefins from synthesis gas: a review [J].ACS Catalysis, 2013, 3(9): 2130-2149.
[8]Luque R, de la Osa A R, Campelo J M, et al.Design and development of catalysts for biomass-to-liquid-Fischer-Tropsch (BTL-FT) processes for biofuels production [J].Energy & Environmental Science, 2012, 5(1): 5186-5202.
[9]Das D, Ravichandran G, Chakrabarty D K.Conversion of syngas to light olefins over silicalite-1 supported iron and cobalt catalysts: effect of manganese addition [J].Catalysis Today, 1997, 36(3): 285-293.
[10]Malessa R, Baerns M.Iron/manganese oxide catalysts for Fischer-Tropsch synthesis (Ⅳ): Activity and selectivity [J].Industrial & Engineering Chemistry Research, 1988, 27(2): 279-283.
[11]Frossling N.The evaporation of falling drops [J].Gerlande Beitr Geophys, 1938, 52: 170-216.
[12]Weisz P, Hicks J.The behaviour of porous catalyst particles in view of internal mass and heat diffusion effects [J].Chemical Engineering Science, 1962, 17(4): 265-275.
[13]Su Junjie(苏俊杰), Mao Wei(茅威), Yang Zhen(杨震), Xu Jing(徐晶), Han Yifan(韩一帆).Kinetics of CoCu/SiO2for synthesis of lower carbon mixed alcohols directly from syngas [J].CIESC Journal(化工学报), 2014, 65(1): 143-151.
[14]Shroff M D, Kalakkad D S, Coulter K E, et al.Activation of precipitated iron Fischer-Tropsch synthesis catalysts [J].Journal of Catalysis, 1995, 156(2): 185-207.
[15]Butt J.Carbide phases on iron-based Fischer-Tropsch synthesis catalysts (Ⅰ): Characterization studies [J].Catalysis Letters, 1990, 7(1-4): 61-81.
[16]Kuivila C, Stair P, Butt J.Compositional aspects of iron Fischer-Tropsch catalysts: an XPS/reaction study [J].Journal of Catalysis, 1989, 118(2): 299-311.
[17]Yang Z, Pan X, Wang J, et al.FeN particles confined inside CNT for light olefin synthesis from syngas: effects of Mn and K additives [J].Catalysis Today, 2012, 186(1): 121-127.
[18]Fierro J L G.Cu-promoted Fe2O3/MgO-based Fischer-Tropsch catalysts of biomass-derived syngas [J].Industrial & Engineering Chemistry Research, 2015, 54(3): 911-921
[19]Tang L, Song C, Li M, et al.Study of K/Mn-MgO supported Fe catalysts with Fe(CO)5and Fe(NO3)3as precursors for CO hydrogenation to light alkenes [J].Chinese Journal of Chemistry, 2013, 31(10): 1263-1268.
[20]Jensen K, Massoth F.Studies on iron-manganese oxide carbon monoxide catalysts (Ⅱ): Carburization and catalytic activity [J].Journal of Catalysis, 1985, 92(1): 109-118.
[21]Mei D, Rousseau R, Kathmann S M, et al.Ethanol synthesis from syngas over Rh-based/SiO2catalysts: a combined experimental and theoretical modeling study [J].Journal of Catalysis, 2010, 271(2): 325-342.
[22]Cheng J, Hu P, Ellis P, et al.A DFT study of the chain growth probability in Fischer-Tropsch synthesis [J].Journal of Catalysis, 2008, 257(1): 221-228.
[23]Cheng J, Hu P, Ellis P, et al.A first-principles study of oxygenates on Co surfaces in Fischer-Tropsch synthesis [J].The Journal of Physical Chemistry C, 2008, 112(25): 9464-9473.
[24]Cheng J, Hu P, Ellis P, et al.Some understanding of Fischer-Tropschsynthesis from density functional theory calculations [J].Topics in Catalysis, 2010, 53(5/6): 326-337.
[25]Cheng J, Song T, Hu P, et al.A density functional theory study of the α-olefin selectivity in Fischer-Tropsch synthesis [J].Journal of Catalysis, 2008, 255(1): 20-28.
[26]Dry M, Shingles T, Boshoff L.Rate of the Fischer-Tropsch reaction over iron catalysts [J].Journal of Catalysis, 1972, 25(1): 99-104.
[27]Yang J, Liu Y, Chang J, et al.Detailed kinetics of Fischer-Tropsch synthesis on an industrial Fe-Mn catalyst [J].Industrial & Engineering Chemistry Research, 2003, 42(21): 5066-5090.
[28]Ahón V R, Costa Jr E F, Monteagudo J E, et al.A comprehensive mathematical model for the Fischer-Tropsch synthesis in well-mixed slurry reactors[J].Chemical Engineering Science, 2005, 60(3): 677-694.
[29]Tsubaki N, Yoshii K, Fujimoto K.Anti-ASF distribution of Fischer-Tropsch hydrocarbons in supercritical-phase reactions [J].Journal of Catalysis, 2002, 207(2): 371-375.
[30]Govender N S, Botes F G, de Croon M H J M, et al.Mechanistic pathway for C2+hydrocarbons over an Fe/K catalyst [J].Journal of Catalysis, 2014, 312: 98-107.
猜你喜欢
大众文艺(2022年16期)2022-09-07
数学物理学报(2022年1期)2022-03-16
农药科学与管理(2019年5期)2019-08-13
华东师范大学学报(自然科学版)(2019年3期)2019-06-24
西安工程大学学报(2016年2期)2016-06-05
当代化工研究(2016年7期)2016-03-20
当代化工研究(2016年2期)2016-03-20
当代化工研究(2016年2期)2016-03-20
化工进展(2015年6期)2015-11-13
中国工程咨询(2015年2期)2015-02-14
