
基于ATR-UV 实时浓度检测的扑热息痛冷却结晶过程研究
2015-08-20 06:16关国强唐凯李红卢帅涛江燕斌
化工学报 2015年9期
关国强,唐凯,李红,卢帅涛,江燕斌
(1 华南理工大学化学与化工学院,广东 广州 510640;2 传热强化与过程节能教育部重点实验室,广东 广州 510640; 3 广东肇庆星湖生物科技股份有限公司技术中心,广东 肇庆 526040)
引 言
结晶是化学工程中重要的单元操作,广泛应用于生物、制药和精细化工等领域[1]。随着高附加值功能性精细化学品的需求增加,传统基于“黑箱模型”的试差结晶过程设计已无法满足现代产品工程的需要。因此,基于过程分析技术(process analytical technology,PAT)的结晶过程控制与优化技术已成为结晶过程研究和应用的热点[2-3]。
结晶是在组分过饱和度驱动下,克服分子有规化“阻力”的液固分离过程,故过饱和度对结晶成核、晶粒生长和团聚有重要的影响。由于过饱和度是组分浓度与相应温度下平衡浓度的函数,为获得形貌优异的晶体产品,需对结晶过程的组分浓度和温度进行实时监控,结合粒度在线检测,进而有效调节结晶过程操作参数。相比传统的“先分离、后检测”的离线浓度监测技术,如高效液相色谱(high performance liquid chromatography,HPLC)等[4],结合衰减全反射技术(attenuated total reflection,ATR)信号采集的浓度检测直接光谱法能实时测定结晶体系局部浓度分布,已成为PAT 浓度实时检测的重要手段。ATR 光谱分析按其光源主要分为红外(infrared spectrum, IR)和紫外(ultraviolet spectrum, UV)分析。相对于采用ATR-IR 的大量报道[5-16],ATR-UV 因侦测距离与波长呈反比,故能分析更薄层表面信息,已成为新近结晶过程研究的有效方法[17]。聚焦光束反射测量技术(focused beam reflectance measurement,FBRM)是结晶过程广泛使用的在线颗粒粒度检测方法[18-19]。FBRM 通过测定弦长分布关联结晶过程的晶粒尺度分布(crystal size distribution,CSD)变化、晶体成核、生长及粒间团聚等现象。采用FBRM已成为当前结晶产品控制的重要手段,其最新应用研究包括作为直接成核控制的控制传感器[20]。
为构建“质量源于设计”(quality by design,QbD)的高效结晶分离系统,本研究在先前UV-vis法[21]研究基础上采用更先进的ATR-UV 方法,通过近年广泛应用于光谱分析的偏最小二乘法(partial least squares,PLS)[22]实现典型活性药物组分——扑热息痛(paracetamol,PCM)乙醇溶液结晶过程的实时浓度分析。进而结合FBRM 实时分析,研究不同降温方式对结晶过程的影响。
1 实验材料和方法
1.1 材料
PCM(CAS#103-90-2,纯度≥99%)购自上海思域化工科技有限公司;分析纯无水乙醇购自国药集团化学试剂有限公司。
1.2 结晶实验系统及主要仪器设备
PCM-乙醇体系冷却结晶实验系统置如图1 所示,它由500 ml 带搅拌夹套结晶釜、程控恒温冷却系统、ATR-UV 检测系统、FBRM 分析系统及计算机数据采集系统组成。主要分析仪器见表1,分别采用ATR-UV检测系统和FBRM实时分析结晶过程浓度和晶体粒度分布(crystal size distribution,CSD),用偏光显微镜观测晶体产品的形貌,其他仪器包括样品处理所需的真空干燥箱和分析天平等。

表1 主要仪器设备一览表Table 1 List of main experimental instruments
1.3 ATR-UV 浓度关联及校验
配制不同浓度的PCM 乙醇溶液,采用ATR-UV光谱仪测定其不同温度下光谱响应(以纯乙醇为空白),见表2。

表2 ATR-UV 浓度关联实验条件Table 2 Experimental parameters for ATR-UV correlation
ATR-UV 光谱数据采用PLS 法进行分析,式(1)用于关联不同温度(T)下PCM 的浓度

式中,Ai为选定的特征波长吸光度值;模型参数a、b 和ci通过PLS 拟合获得。
采用Umetrics 公司的SIMCA-P 11.5 分析软件导入对已知浓度的PCM 溶液在不同温度下测定的特征波长吸光度值,采用压缩-中心化方法进行数据标准化后,根据霍特林T2(Hotellin T2,置信水平为0.05)分布检验数据点并建立回归建模用的数据集。以最小差均方根平均偏差(root-mean-square error,RMSE)为目标函数,如式(2)所示,通过PLS 回归式中的模型参数,使RMSE 最小。

式中,yi和ˆiy 分别为训练集和校正集的浓度;n为数据点数目。
对建立的ATR-UV 在线PCM 浓度检测模型,采用重量法进行校验。重量法浓度测定过程如下[23-24]:
(1)取样:采用预热的带砂芯过滤头1 ml 注射器实时取样,其中平均孔径为1 μm;
(2)真空干燥:将溶液迅速移至称量瓶并置于真空干燥箱中(40℃,40 kPa)干燥24 h 至恒重;
(3)称量:在电子分析天平上测定样品残留固体质量。
PCM 在乙醇中的浓度(y),用式(3)计算

式中,WR为样品残留固体质量;WS为取样 质量。
1.4 饱和溶解度关联及过饱和度测定
采用文献[25]报道的PCM 溶解度数据(y∗),结合实时浓度检测结果,即可计算结晶过程的关键过饱和度(S),如式(4)所示

1.5 PCM 冷却结晶实验
应用上述ATR-UV 浓度关联可实时分析PCM结晶中的过饱和度变化,通过同步调节过程温度,实现受控结晶。为此在如图1 所示结晶系统中进行PCM 结晶实验。
实验具体步骤如下:
(1)母液配制:称量85.6 g PCM 置于结晶釜中,加入300 ml 乙醇溶剂,维持搅拌200 r·min-1,60℃条件下恒温1 h 以确保固体颗粒完全溶解;
(2)加入晶种:待母液冷却至T0(=48℃)时加入4.7 g 13~21 μm 的晶种并稳定10 min;
(3)冷却结晶:通过程序控制恒温槽调控结晶温度变化,使结晶釜内PCM 溶液冷却结晶,同时采用ATR-UV 关联溶液的过饱和度和通过FBRM分析结晶产物CSD,冷却结晶持续时间(t1)为3 h。
分别采用3 种冷却结晶温控策略:自然冷却(natural cooling policy,NCP)、线性冷却(linear cooling policy,LCP)和受控冷却(controlled cooling policy,CCP)。进行NCP 结晶时,维持结晶釜夹套中循环水温度为固定温度;LCP 和CCP 结晶时,控制结晶釜夹套中循环水温度使釜内PCM 溶液温度按式(5)冷却至T1(=5℃)。
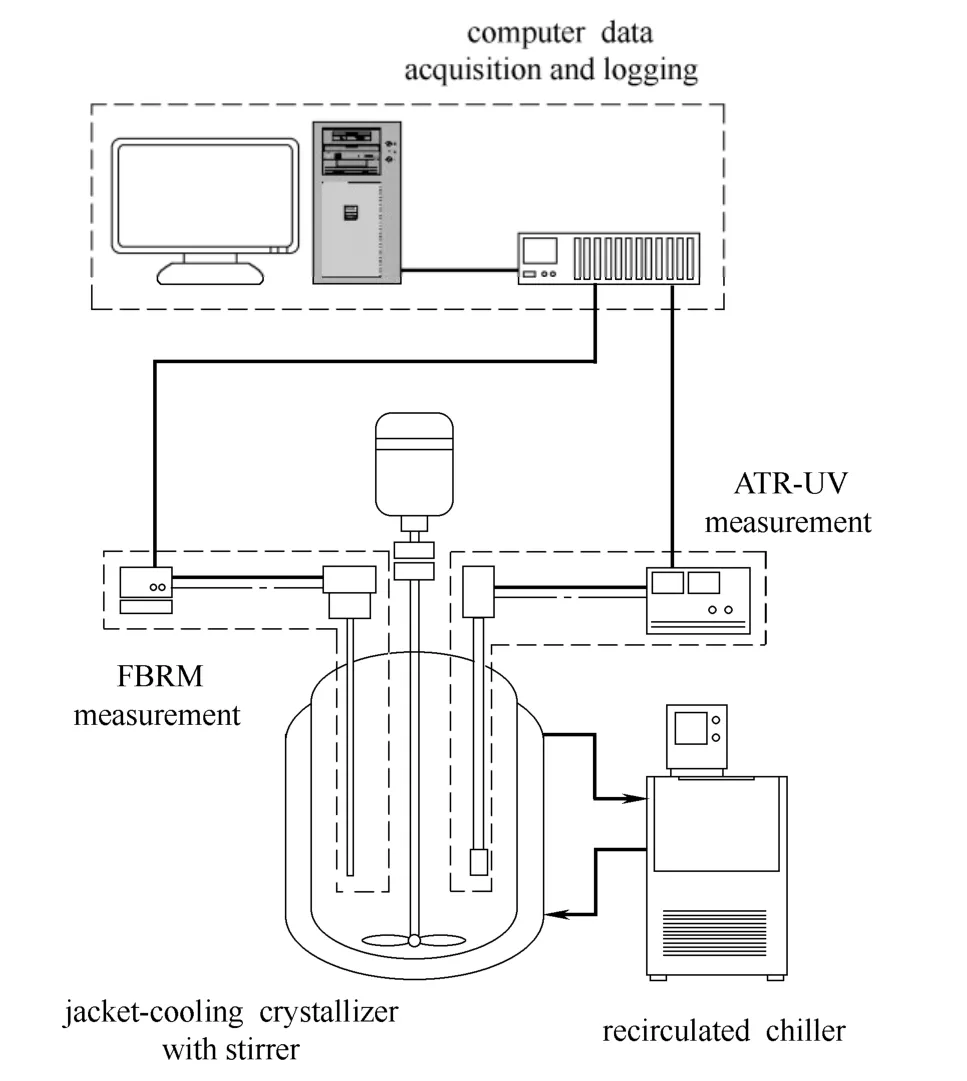
图1 PCM 结晶ATR-UV 和FBRM 实时监控实验系统Fig.1 Schematic of PCM crystallization using ATR-UV and FBRM real-time measurement

式中,量纲1 时间(τ)和温度(Θ)分别为

2 实验结果与讨论
2.1 ATR-UV 实时浓度检测模型的建立和校验
图2、图3 分别为不同温度和浓度的PCM 乙醇溶液ATR-UV 谱图。由图可知,PCM 乙醇溶液的ATR-UV 谱图在波长(λ)255 μm 附近存在特征吸收峰。图2 表明,当溶液温度由60℃降低至40℃时,特征吸收峰位置由255.14 μm 变化至256.42 μm,可见温度对ATR-UV 特征吸收峰位置的影响较小,故ATR-UV 实时浓度测定中可不对温度进行校正。图3 表明,当PCM 浓度从0.1686 g·g-1乙醇提高至0.3844 g·g-1乙醇时,特征吸收峰位置由251.09 μm红移至256.42 μm。相对于温度,PCM 浓度增加使特征吸收峰位置发生略微的红移。

图2 PCM 乙醇溶液在不同温度下的ATR-UV 谱图Fig.2 ATR-UV spectra of PCM in ethanol for varied temperatures

图3 不同浓度PCM 乙醇溶液的ATR-UV 谱图Fig.3 ATR-UV spectra of PCM in ethanol for varied concentrations

表3 PCM 在乙醇中的ATR-UV 浓度关联参数Table 3 Parameters for correlating concentration of PCM in ethanol using ATR-UV spectra
为有效关联不同温度和浓度下PCM 乙醇溶液的ATR-UV 响应,用式(1)对包含操作范围中特征峰位置范围的信息进行关联。通过如1.3 节所述方法,采用PLS 法关联PCM 浓度,各参数关联结果见表3,其RMSE 为0.0019。
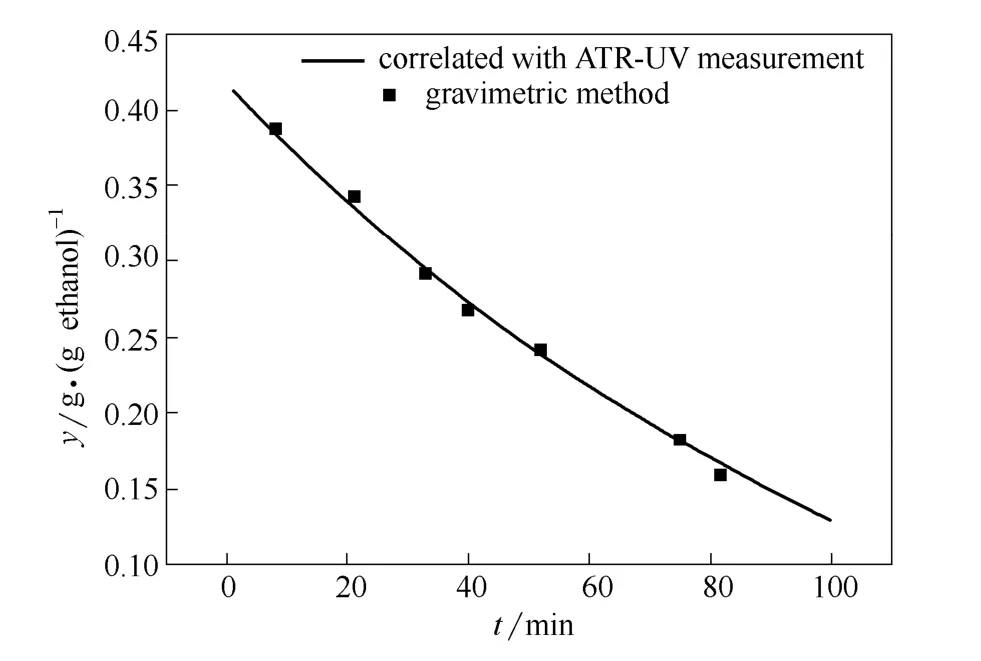
图4 ATR-UV实时浓度检测结果与重量法浓度测定值的比较Fig.4 Comparison of PCM concentration measured by ATR-UV method and gravimetric method
在PCM 实际结晶操作中应用ATR-UV 进行实时浓度检测,并在结晶开始后不同时间(例如8、21、33、40、52、75 和82 min)抽取少量的PCM乙醇溶液采用重量法测定其浓度,ATR-UV 测定结 果与重量法测定值的对比如图4 所示。图4 表明,采用ATR-UV 实时关联的PCM 浓度与重量法测定结果一致,其RMSE 为0.0075,说明1.3 节所述建立的ATR-UV 浓度关联方法可实现结晶过程PCM浓度的实时检测。
2.2 温度控制策略对PCM 冷却结晶的影响
2.2.1 冷却结晶温控策略对晶粒产品形貌的影响 本文通过调节结晶釜夹套内循环水温度变化实现对结晶温度的控制,研究了NCP、LCP 和CCP 3 种冷却结晶温度控制策略对结晶产品质量的影响。PCM在乙醇中的结晶实验如1.5 节所述,其中冷却时间为180 min,终点为5℃。为便于比较温度和浓度的变化,对不同时间的温度及浓度进行了量纲1 化。3种不同的降温方式结晶温度和浓度随时间的变化见图5,其中PCM 的浓度采用ATR-UV 实时测定。
对于NCP 控温,图5 表明,由于结晶过程固含量较低,结晶热效应不显著,故NCP 的结晶温度变化曲线近似于自然冷却过程的温度变化曲线,即:结晶初段系统与环境温差最大而使温度快速下降,随后因与环境温差的减少而降温速率降低;结晶浓度曲线形状与温度曲线相似,即结晶初期浓度变化较快,随后浓度变化逐步减少。这是由于结晶初期小粒径晶粒数目较多有利于晶粒加速生长,从而加速了组分在液相中的消耗。随着晶粒尺寸的增加,晶粒生长速度降低,同时NCP 中新晶粒的产生并不显著,故组分浓度变化随结晶进行而减少。
对于LCP 控温,图5 表明,体系的温度变化具有较好的线性,表明本文采用的夹套温控方案能较好地实现结晶过程温度控制。在LCP 中,结晶前期的温度变化比NCP 小,同时由于LCP 的温度呈线性下降,LCP 结晶中段的温度变化比NCP 更显著。由图5 可知,相对于NCP 过程,LCP 在结晶初期的PCM 浓度变化较小,表明LCP 结晶初段晶体成核及生长速度较NCP 慢;随后量纲1 时间τ>0.3(即结晶约60 min 后)时,LCP 浓度变化较逐渐比NCP 显著,表明结晶中后期LCP 的结晶速度较 NCP 快。
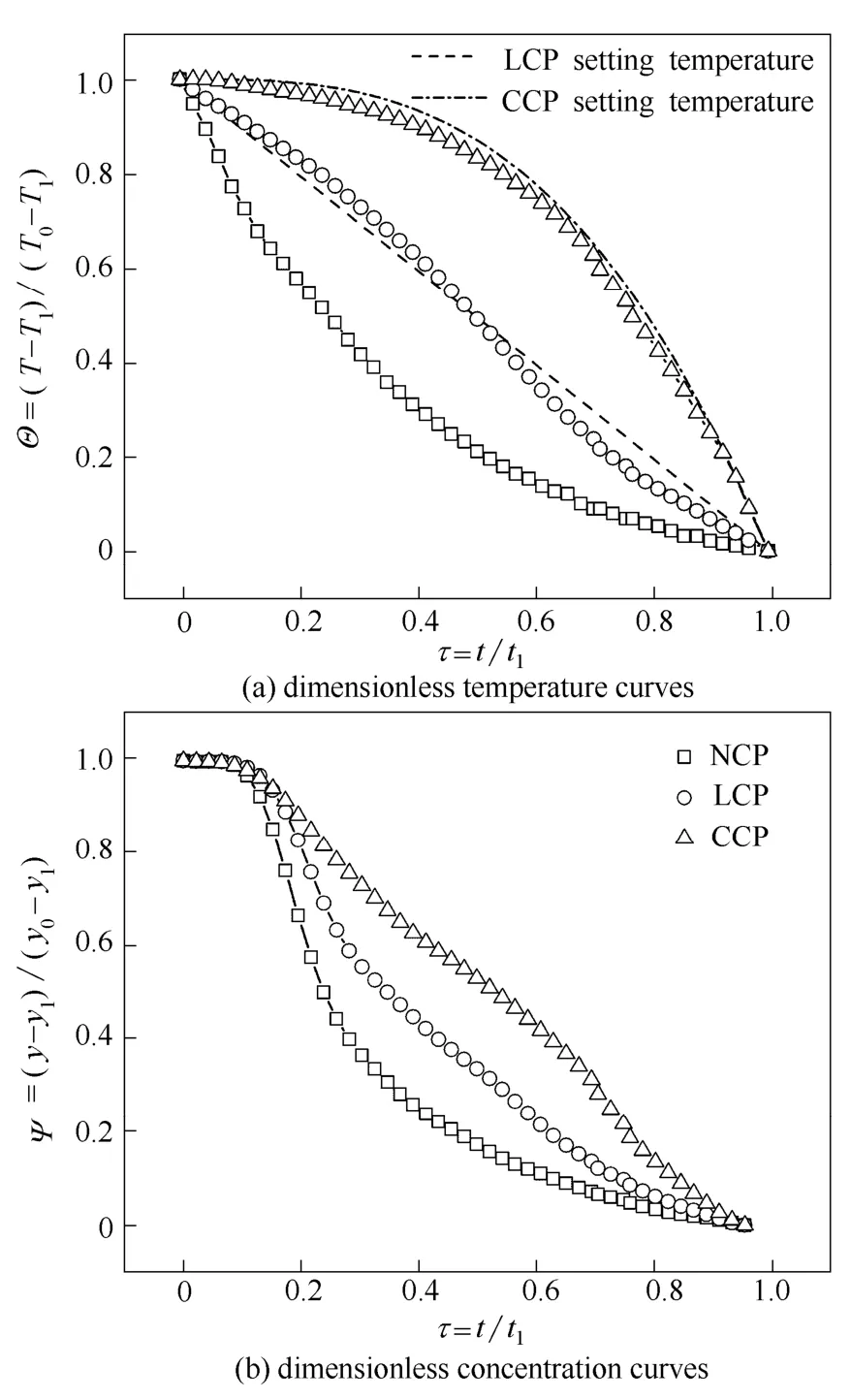
图5 不同降温方式PCM 结晶量纲1 温度和浓度曲线Fig.5 Dimensionless temperature (a) and concentration (b) curves of PCM crystallization
相对于LCP 的线性温度下降,采用CCP 结晶过程的温度变化呈现为先缓慢下降、后加速减少的变化趋势,这种变化与NCP 过程截然相反。同时CCP 过程的浓度变化更为均匀,如图5 所示,在3种冷却策略中,CCP 过程的体系浓度演变曲线具有较好的线性,表明CCP 过程的结晶速率比较稳定。采用不同降温策略所得的晶体产品形貌见图6。偏光显微镜观测发现NCP 和LCP 过程结晶产物的晶粒尺寸都较小,如图6(a)所示,NCP 过程小尺寸晶粒出现了团聚现象;而LCP 中晶粒分散性较NCP有所改善,但发现晶粒尺寸变化较大,例如在图6(b)中大颗粒晶体的尺寸比小颗粒大了5 倍以上。在3 种降温策略中,采用CCP 过程的结晶产品具有 最大的晶粒尺寸和较一致的晶粒尺寸分布,如图6(c)所示,由此说明CCP 是较好的结晶温度控制方式。为进一步阐明CCP 能获得更大、更均一晶体产品的原因,本文分别从过饱和度变化及晶粒CSD演变两个方面深入说明降温方式对结晶过程的影响。

图6 不同降温方式下PCM 冷却结晶产品的显微照片Fig.6 Micropolariscopy of produced PCM crystals using different cooling policy
2.2.2 降温方式对结晶过程过饱和度变化影响 对特定体系的结晶过程,过饱和度是过程的推动力,其大小直接决定了结晶速度的快慢,故对结晶过程过饱和度变化的分析有助于从根本上理解结晶现象。借助ATR-UV 实时浓度关联,可获得不同降温方式的过饱和度随时间变化,如图7 所示。
对于NCP 结晶过程,过饱和度在结晶开始约30 min 内迅速增加,而此时体系的浓度变化不大(如图5 所示),表明此时可能处于PCM 结晶始发的诱导期;由于在高过饱和条件下可能引发晶体高速成核和生长,即类似“爆发结晶”过程,故过饱和度一旦突破其阈值将快速下降,因而在结晶后30~60 min 发现过饱和度急速下降,同时由于溶质在结晶过程的消耗导致了浓度显著下降,这与NCP 浓度变化的测定结果一致。结晶约60 min 后的过饱和度由峰值的1.30 降低至1.10,这时结晶过程因较低的推动力而缓慢进行。

图7 不同降温方式下PCM 冷却结晶过饱和度曲线Fig.7 Supersaturation curves of PCM crystallization using different cooling policy
相对于NCP,LCP前期同样出现了过饱和度“快速增加然后迅速下降”的现象,这表明LCP 可能存在同样的类似“爆发结晶”的过程。然而相对于NCP,LCP 的峰值过饱和度较低,这可能是由于结晶前期LCP 温度变化比NCP 略小所致(如2.2.1 节所述),同时也正是LCP 在结晶后期具有比NCP 更大的温度下降,导致了饱和浓度减少而使结晶约30 min 后的LCP 过饱和度比NCP 过程更高。
显然NCP 和LCP 结晶前期出现的高过饱和度条件所产生的类似“爆发结晶”现象会产生大量的小尺寸颗粒,由此导致了图6(a)、(b)所示较小的产品晶粒尺寸。此外,随着结晶的进行,NCP 较LCP 因具有较小的温度扰动和过饱和度变化而有利于晶粒团聚生长。
相比NCP 和LCP 结晶,CCP 过程过饱和度变化更为温和,图7 表明,CCP 结晶的过饱和度在1.08~1.13 范围变化。特别是在结晶初期CCP 的温度变化较小,由此使过饱和度维持在相对较低(S=1.13)的水平,这能有效地抑制NCP 和LCP出现的类似“爆发结晶”现象,从而有利于晶体的生长,获得粒径较大、分布较窄和晶体形貌较好的产品,如图6(c)所示。
2.2.3 降温方式对结晶产品CSD 的影响 进一步采用FBRM 实时分析不同降温方式的晶粒CSD 变化,以其非加权结果识别晶粒的绝对量变化,结果如图8 所示。由图8 可知:(1)NCP 过程大部分晶颗是在0~30 min 内生成,此后晶粒数目(即实线CSD 曲线下的面积)随时间增加而略微减少,而CSD 分布则变宽。这说明NCP 过程在结晶初期可能存在类似“爆发式结晶”现象,同时NCP 中存在严重的晶粒团聚现象,这使FBRM 将多个晶粒团聚体识别为单一大晶粒,从而测得晶粒数目随时间而减少;(2)LCP 过程的CSD 曲线以点划线表示,LCP在30 min 时晶粒数目因过饱和度较低而比NCP 少,随后因LCP 具有较高的过饱和度不断地成核结晶,由此使晶粒数目随时间增加而增大,这与图7 的结果一致。LCP 过程的CSD 曲线也表明晶体产品的粒度较NCP 的更小,这与图6 观察结果一致;(3)CCP 过程的CSD(如图8 虚线所示)曲线下部面积随时间推移而稳定增加,在0.5 h 时CCP 结晶过程颗粒数目明显低于NCP 和LCP 结晶过程,随着结晶过程的进行,CCP 结晶过程中晶体成核与生长稳定进行,使CSD 向平均粒径增大方向移动且峰高增大,最终产品具有较大晶粒尺寸(与NCP 多晶粒团聚结果相当,而明显大于LCP 晶粒尺寸)和更多的晶粒数量,同时CSD 分布也较NCP 和LCP 更窄。

图8 不同降温方式下PCM 冷却结晶晶粒尺寸分布随时间的变化Fig.8 Crystal size distribution in PCM crystallization using different cooling policy
不同降温方式晶粒体积平均尺寸(d43)随时间的变化如图9 所示。由图8、图9 可知,NCP 结晶过程平均粒径先增长后逐渐趋于稳定在较高水平,属于小数目大粒径分布;LCP 过程晶粒尺寸先增加,后因过饱和度较高成核增加而产生较多小尺寸晶粒,从而导致平均粒径减小;CCP 结晶过程晶体尺寸生长较NCP 和LCP 更为稳定,其最终产品平均粒径也最大,约为130 μm。

图9 不同降温方式下平均粒径随时间的变化Fig.9 Averaged size in PCM crystallization using different cooling policy
3 结 论
(1)基于ATR-UV 测量,采用偏最小二乘法进行浓度关联,成功实现了乙醇-扑热息痛体系结晶过程的实时浓度检测,采用重量法对实时浓度检测结果进行了校验,其均方根平均偏差为0.0075。
(2)采用ATR-UV 实时浓度分析方法,考察了不同降温方法(自然冷却、线性冷却和受控冷却)对乙醇中扑热息痛结晶过程的影响。结合结晶过饱和度的实时分析和FBRM 实时晶粒尺寸测量结果,发现受控冷却结晶过程中晶体成核与生长能在更适宜获得更大、更均一粒子的良性平衡中进行。
(3)采用实时过饱和度分析可建立基于过程分析的优化结晶控制系统,以实现质量源于设计的结晶产品调控。
符 号 说 明
A,B,C,D ——饱和溶解度关联参数
Ai——吸光度
a, b, ci——ATR-UV 浓度关联参数
d43——体积平均粒径,μm
L ——晶粒尺寸,μm
n ——数据点数目
RMSE ——均方根平均偏差
S ——过饱和度
T ——温度,℃
t ——时间,min
W ——样品质量,kg
y ——质量浓度,g/g 溶剂
Θ ——量纲1 温度
λ ——波长,μm
τ ——量纲1 时间
Ψ ——量纲1 浓度
上角标
* ——平衡浓度
下角标
R ——残留量
S ——取样量
0 ——结晶开始
1 ——结晶结束
* ——平衡浓度
[1]Wang Jingkang (王静康).Crystallization (结晶)//Shi Jun (时钧), Wang Jiading (汪家鼎), Yu Guocong (余国琮), Chen Minheng (陈敏恒).Handbook of Chemical Engineering (化学工程手册) [M].Beijing: Chemical Industry Press, 1996.
[2]Yu L X, Lionberger R A, Raw A S, D'Costa R, Wu H, Hussain A S.Applications of process analytical technology to crystallization processes [J].Advanced Drug Delivery Reviews, 2004, 56 (3): 349-369.
[3]Simon L L, Pataki H, Marosi G, Meemken F, Hungerbühler K, Baiker A, Tummala S, Glennon B, Kuentz M, Steele G, Kramer H J M, Rydzak J W, Chen Z, Morris J, Kjell F, Singh R, Gani R, Gernaey K V, Louhi-Kultanen M, O’Reilly J, Sandler N, Antikainen O, Yliruusi J, Frohberg P, Ulrich J, Braatz R D, Leyssens T, von Stosch M, Oliveira R, Tan R B H, Wu H, Khan M, O’Grady D, Pandey A, Westra R, Delle-Case E, Pape D, Angelosante D, Maret Y, Steiger O, Lenner M, Abbou-Oucherif K, Nagy Z K, Litster J D, Kamaraju V K, Chiu M-S.Assessment of recent process analytical technology (PAT) trends: a multiauthor review [J].Organic Process Research & Development, 2015, 19 (1): 3-62.
[4]Palablylk i M, Onur F.The simultaneous determination of phenylephrine hydrochloride, paracetamol, chlorpheniramine maleate and dextromethorphan hydrobromide in pharmaceutical preparations [J].Chromatographia, 2007, 66 (1): 93-96.
[5]Cornel J, Mazzotti M.Estimating crystal growth rates using in situ ATR-FTIR and Raman spectroscopy in a calibration-free manner [J].Industrial & Engineering Chemistry Research, 2009, 48 (23):10740-10745.
[6]Duffy D, Barrett M, Glennon B.Novel, calibration-free strategies for supersaturation control in antisolvent crystallization processes [J].Crystal Growth & Design, 2013, 13 (8): 3321-3332.
[7]Frawley P J, Mitchell N A, O'Ciardha C T, Hutton K W.The effects of supersaturation, temperature, agitation and seed surface area on the secondary nucleation of paracetamol in ethanol solutions [J].Chemical Engineering Science, 2012, 75: 183-197.
[8]Hermanto M W, Chow P S, Tan R B H.Operating strategy to produce consistent CSD in combined antisolvent-cooling crystallization using FBRM [J].Industrial & Engineering Chemistry Research, 2012, 51 (42): 13773-13783.
[9]Mitchell N A, O'Ciardha C T, Frawley P J.Estimation of the growth kinetics for the cooling crystallisation of paracetamol and ethanol solutions [J].Journal of Crystal Growth, 2011, 328 (1): 39-49.
[10]Nagy Z K, Chew J W, Fujiwara M, Braatz R D.Comparative performance of concentration and temperature controlled batch crystallizations [J].Journal of Process Control, 2008, 18 (3/4): 399-407.
[11]Nagy Z K, Fujiwara M, Woo X Y, Braatz R D.Determination of the kinetic parameters for the crystallization of paracetamol from water using metastable zone width experiments [J].Industrial & Engineering Chemistry Research, 2008, 47 (4): 1245- 1252.
[12]O'Ciardha C T, Mitchell N A, Hutton K W, Frawley P J.Determination of the crystal growth rate of paracetamol as a function of solvent composition [J].Industrial & Engineering Chemistry Research, 2012, 51 (12): 4731-4740.
[13]Trifkovic M, Sheikhzadeh M, Rohani S.Kinetics estimation and single and multi-objective optimization of a seeded, anti-solvent, isothermal batch crystallizer [J].Industrial & Engineering Chemistry Research, 2008, 47 (5): 1586-1595.
[14]Trifkovic M, Sheikhzadeh M, Rohani S.Multivariable real-time optimal control of a cooling and antisolvent semibatch crystallization process [J].AIChE Journal, 2009, 55 (10): 2591-2602.
[15]Trifkovic M, Sheikhzadeh M, Rohani S.Determination of metastable zone width for combined anti-solvent/cooling crystallization [J].Journal of Crystal Growth, 2009, 311 (14): 3640-3650.
[16]Yu Z Q, Chow P S, Tan R B H.Application of attenuated total reflectance-Fourier transform infrared (ATR-FTIR) technique in the monitoring and control of anti-solvent crystallization [J].Industrial & Engineering Chemistry Research, 2006, 45 (1): 438-444.
[17]Saleemi A N, Rielly C D, Nagy Z K.Comparative investigation of supersaturation and automated direct nucleation control of crystal size distributions using ATR-UV/vis spectroscopy and FBRM [J].Crystal Growth & Design, 2012, 12 (4): 1792-1807.
[18]Doki N, Seki H, Takano K, Asatani H, Yokota M, Kubota N.Process control of seeded batch cooling Crystallization of the metastable α-Form glycine using an in-situ ATR-FTIR spectrometer and an in-situ FBRM particle counter [J].Crystal Growth & Design, 2004, 4 (5): 949-953.
[19]Sheikhzadeh M, Trifkovic M, Rohani S.Real-time optimal control of an anti-solvent isothermal semi-batch crystallization process [J].Chemical Engineering Science, 2008, 63 (3): 829-839.
[20]Abu Bakar M R, Nagy Z K, Saleemi A N, Rielly C D.The impact of direct nucleation control on crystal size distribution in pharmaceutical crystallization processes [J].Crystal Growth & Design, 2009, 9 (3): 1378-1384.
[21]Zhang Y, Jiang Y B, Zhang D K, Li K X, Qian Y.On-line concentration measurement for anti-solvent crystallization of β-artemether using UV-vis fiber spectroscopy [J].Journal of Crystal Growth, 2011, 314 (1): 185-189.
[22]Zhang R, Ma J, Li J, Jiang Y B, Zheng M Y.Effect of pH, temperature and solvent mole ratio on solubility of disodium 5-guanylate in water + ethanol system [J].Fluid Phase Equilibria, 2011, 303 (1): 35-39.
[23]Zhang Y, Jiang Y B, Li K X, Qian Y.Solubility of β-artemether in methanol + water and ethanol + water from (288.85 to 331.95) K [J].Journal of Chemical Engineering Data, 2009, 54 (4): 1340-1342.
[24]Samadi-Maybodi A, Hassani Nejad-Darzi S K.Simultaneous determination of paracetamol, phenylephrine hydrochloride and chlorpheniramine maleate in pharmaceutical preparations using multivariate calibration 1 [J].Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2010, 75 (4): 1270-1274.
[25]Worlitschek J, Mazzotti M.Model-based optimization of particle size distribution in batch-cooling crystallization of paracetamol [J].Crystal Growth & Design, 2004, 4 (5): 891-903.
猜你喜欢
学与玩(2022年12期)2023-01-11
小资CHIC!ELEGANCE(2021年25期)2021-07-29
小哥白尼(神奇星球)(2020年7期)2021-01-18
老友(2017年7期)2017-08-22
能源(2016年1期)2016-12-01
中国塑料(2016年9期)2016-06-13
少儿科学周刊·儿童版(2015年7期)2015-11-24
中国塑料(2015年7期)2015-10-14
理科考试研究·高中(2014年11期)2014-11-26
小天使·一年级语数英综合(2014年8期)2014-06-26
