
固相萃取净化-气相色谱质谱法测定茶叶中22种禁用农药残留量
2015-08-11 09:53陆小磊蒋美蓉叶美君周卫龙
中国茶叶加工 2015年1期
陆小磊,蒋美蓉,叶美君,周卫龙
(1.中华全国供销合作总社杭州茶叶研究院,国家茶叶质量监督检验中心,浙江杭州 310016;2.浙江经贸职业技术学院,浙江杭州,310018)
固相萃取净化-气相色谱质谱法测定茶叶中22种禁用农药残留量
陆小磊1,蒋美蓉2,叶美君1,周卫龙1
(1.中华全国供销合作总社杭州茶叶研究院,国家茶叶质量监督检验中心,浙江杭州 310016;
2.浙江经贸职业技术学院,浙江杭州,310018)
摘要:建立了测定茶叶中22种禁用农药残留量的气相色谱-质谱联用分析方法。茶叶样品经乙腈均质提取后,经石墨化碳-氨基复合固相萃取小柱净化后,采用气相色谱质谱联用仪在选择离子监测模式下进行定性定量。结果表明,22种农药在0.05~1.00 μg/mL范围内的标准工作曲线相关系数大于0.99;在1.00、0.25和0.025 mg/kg浓度添加水平下,22种农药的回收率在65%~120%之间,相对标准偏差(RSDs)小于10%;方法检出限为0.001~0.031 mg/kg。该方法准确、快速、稳定,适合实际检测需要。
关键词:茶叶;禁用农药;残留量;固相萃取;气相色谱质谱法
中国茶叶加工2015(1):11~17
农药作为一项茶园生产过程中的重要投入品,对有效防治茶园病虫害、增加茶叶产量、提高茶叶品质起到重要作用。但是随着对农药性质、环境行为和生物安全性的深入研究,有些常用农药具有不易降解、致畸、致癌等特点,长期施用该类农药,将会严重污染环境和危害人体健康[1]。为此,我国农业部发布194、199、274公告文件,列出禁用农药和限用农药清单,其中包括23种全面禁止使用的农药,16种不得在茶树上使用的农药[2-3]。同时,国家卫计委对GB 2763《食品中农药最大残留限量》进行不断修订,计划把所有禁用农药残留限量列入国家食品强制性标准中[4]。因此,有必要建立一种准确、快速、可靠的茶叶中禁用农药多残留检测方法。
目前对禁用农药的检测手段主要有气相色谱法(GC)、液相色谱法(LC)、色谱-串联质谱法[5-8]等,但针对茶叶中禁用农药测定的研究不多。茶叶中含有大量不确定的干扰物,传统的分析方法净化效果不佳,本文采用均质提取和固相萃取(SPE)净化,用气相色谱-质谱联用仪(GC-MS)进行测定,能够准确地对22种有机磷、有机氯和氨基甲酸酯类禁用农药进行定性定量。
1 材料与方法
1.1仪器与试剂
主要仪器:Agilent 7890A-5975C气相色谱-质谱联用仪 (GC-MS),配有电子轰击离子源(Electron impact ion source,EI),ChemStation质谱工作站(美国 Agilent公司);分析天平(感量0.01 mg和0.1 mg,瑞士METTLER TOLEDO公司);SC-3610低速离心机 (安徽中科中佳科学仪器有限公司);T10高速组织分散机 (德国IKA公司),最大转速30000 rpm;VM06型6位大体积负压SPE装置 (天津Agela公司);CS501旋转蒸发仪(上海瑞立科学仪器有限公司);MTN-2800D氮吹浓缩装置(天津奥特赛恩斯仪器有限公司)。
试剂:乙腈(色谱纯,美国TEDIA公司);丙酮,乙酸乙酯,石油醚(分析纯,华东医药股份有限公司);CNW 50 mL塑料螺纹具塞离心管(上海安谱科学仪器有限公司);200 mL圆底烧瓶,5 mL 和10 mL刻度移液管 (天津市天玻玻璃仪器有限公司);GC-e/NH2500 mg/6 mL WondaSep SPE小柱(岛津技迩商贸有限公司);2 mL一次性无菌注射器 (杭州龙德医用器械有限公司);0.45 μm微孔过滤膜(尼龙,天津Agela公司)。混合标准溶液,包括涕灭威、克百威、内吸磷、灭线磷、杀虫脒、治螟磷、久效磷、甲拌磷、特丁硫磷、地虫硫磷、氯唑磷、磷胺、甲基对硫磷、艾氏剂、三氯杀螨醇、对硫磷、甲基异柳磷、硫环磷、苯线磷、狄氏剂、除草醚、蝇毒磷等22种农药(100.0 μg/mL,1.5 mL,美国AccuStandard公司);内标物质:环氧七氯(纯度100%,美国AccuStandard公司)。
1.2方法
1.2.1标准溶液配置
准确称取约5 mg(精确到0.01 mg)环氧七氯至50 mL容量瓶中,用丙酮溶解定容,再稀释配置成浓度为2.10 μg/mL的内标溶液。
用2 mL样品空白溶液和一定体积的混合标准溶液配置成浓度为 0.05、0.10、0.20、0.50、1.00 μg/mL的基质混合标准工作溶液,并加入40 μL内标溶液。基质混合标准工作溶液现用现配。
1.2.2样品制备
取茶叶约500 g放入粉碎机中磨碎,制备供分析样品,并放置于避光阴凉干燥处保存。
1.2.3样品前处理
(a)提取。称取2 g试样(精确至0.0001 g)于50 mL具塞离心管中,加入10 mL乙腈,12000 rpm均质1 min,3500 rpm离心后移取提取液,残渣再用10 mL乙腈重复均质提取一次,合并提取液于200 mL圆底烧瓶中,于40℃水浴中减压浓缩至干,待净化。
(b)净化。将5 mL淋洗液(乙腈+丙酮+乙酸乙酯=3+2+1,体积比)以约每秒1滴的速度通过WondaSep SPE小柱,并保持柱子湿润,弃去;然后用5 mL淋洗液对浓缩后的净化液洗涤3次并加入SPE小柱,以约每秒1滴的速度通过小柱,并收集于200 mL圆底烧瓶中;再用20 mL淋洗液(乙腈+丙酮+乙酸乙酯=3+2+1,体积比)淋洗小柱,合并收集洗脱液。
(c)定容。将洗脱液在40℃下减压浓缩近干,吹干,用石油醚准确定容至2 mL,加入40 μL内标溶液,供GC-MS检测。
1.2.4添加回收率试验
称取经测定不含目标农药的茶叶空白样品2 g(精确至0.0001 g),分别添加不同浓度混合标准溶液,使其相当于1.00、0.25、0.025 mg/kg浓度添加水平,混匀后,按照1.2.3进行处理提取与净化,每个浓度水平重复5次,设置1个空白样品对照;移取相当量混合标准溶液至进样小瓶中,加入按照1.2.3提取后的空白茶叶样品溶液稀释定容,加入40 μL内标溶液,作为基质标准进行测定,计算添加回收率、相对标准偏差和检出限。
1.2.5气相色谱-质谱条件
色谱柱:DB-5 MS石英毛细管柱 (30 m× 0.25 mm×0.25 μm);载气:氦气 (He),纯度≥99.999%,流速1.0 mL/min;进样口温度:290℃;进样量:1 μL,不分流进样;升温程序:40℃保持1 min,然后以30℃/min程序升温至130℃,再以5℃/min升温至250℃,再以10℃/min升温至300℃,保持5 min。电离方式:电子轰击电离源(EI)70 eV;接口温度:280℃;四级杆温度:150℃;离子源温度:230℃;溶剂延迟:5 min。
2 结果与讨论
2.1前处理方法的优化
在提取方式的选择上,本试验采用了均质的提取方式,即在茶叶中加入一定量溶剂作为提取剂,通过高速均质的方式,提高茶叶破碎度,使得茶叶基质与提取溶剂充分接触,提高农药的提取效率。与索氏提取、超声提取等传统提取方式相比,均质提取具有操作简单、快速的特点。因此,本文选用均质提取方式。
在提取溶剂的选择上,由于目标农药包括有机磷、有机氯和氨基甲酸酯类化合物,这三种农药易溶于有机溶剂,乙腈、二氯甲烷、石油醚均为良好的提取剂,但考虑到茶叶基质较多,而乙腈提取出的色素、油脂类杂质较少,且能够沉淀蛋白质等杂质[8],因此,本试验选取乙腈作为提取溶剂。
虽然采用乙腈提取可以选择性地除去一部分茶叶基质,但是还需做进一步的净化处理以除去剩余的色素和其他小分子杂质。石墨化碳能很好地吸附色素,氨基能有效地吸附有机酸和糖分,同时使用两种填料进行净化能够很好地除去大部分杂质[8]。
考虑到目标化合物的极性不一,但均能溶于乙腈,因此选择乙腈。根据GB/T 23204标准,洗脱液为乙腈+甲苯+乙酸(75+25+1,体积比),因甲苯毒性较大,且较难获得,因此选择一定比例极性较强的丙酮和极性较弱的乙酸乙酯来调节洗脱效率。在洗脱液的选择上,通过馏分试验(图1),可以看出使用乙腈+丙酮+乙酸乙酯(3+2+1,体积比)可以使目标化合物在洗脱量5~15 mL之间被洗出,考虑到该方法需要对更多的禁用农药进行测定,个别农药可能在15 mL的洗脱量下不能被完全洗脱,因此试验中采用20 mL的乙腈+丙酮+乙酸乙酯(3+ 2+1,体积比)来洗脱小柱中的目标化合物。
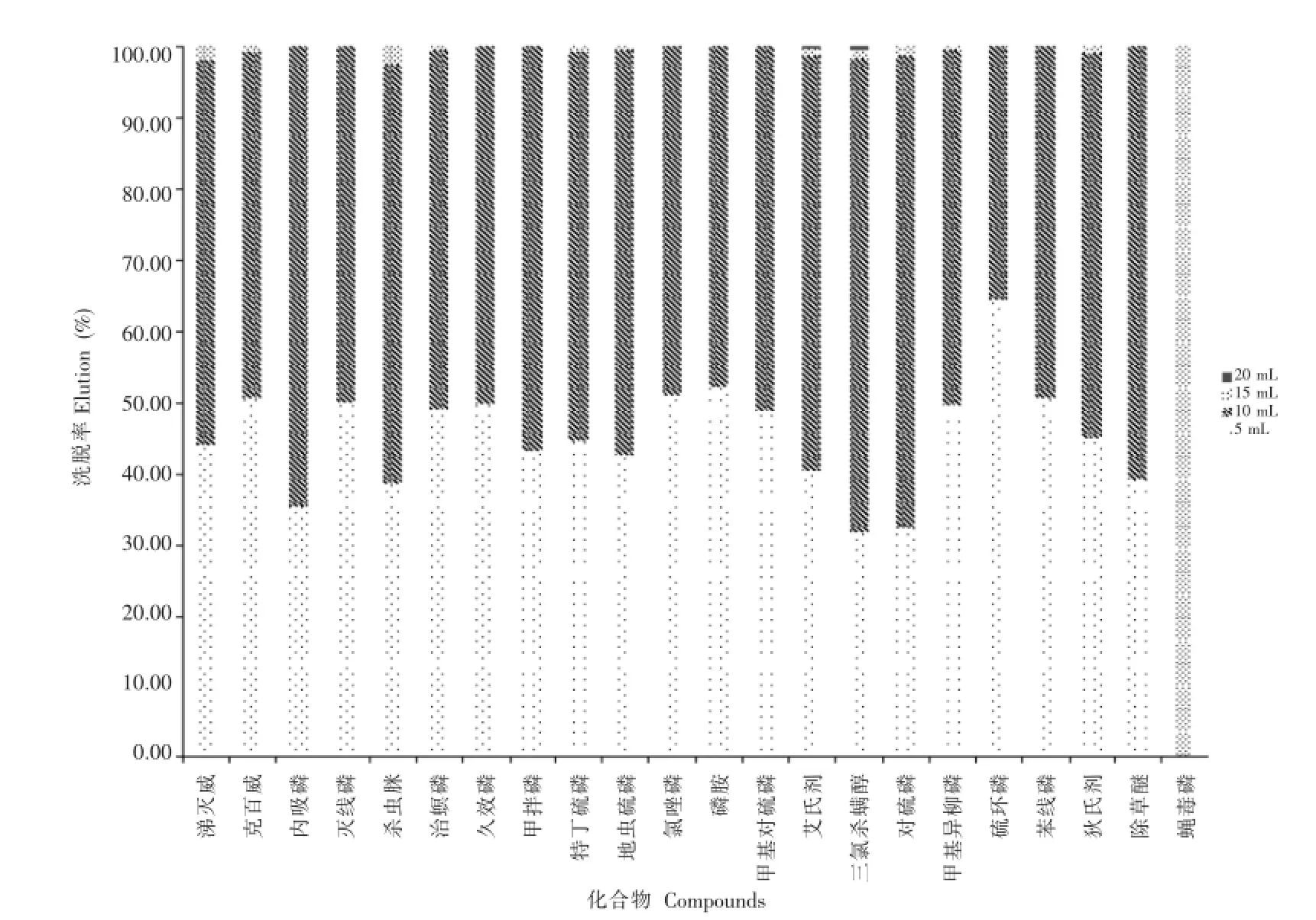
图1 乙腈+丙酮+乙酸乙酯(3+2+1,体积比)对22种农药的洗脱效果Fig.1 Elution rates of 22 pesticides in Carb-NH2with acetonitrile+acetone+ethyl acetate(3+2+1,volume ratio)
2.2气相色谱-质谱条件优化
石英毛细管柱DB-5 MS作为一款高性能通用色谱柱,不仅对大部分农药化合物有很好的保留特性,而且其低流失的特点能够减少噪音,提高信噪比。采用不同的升温速率能够有效分离目标化合物,并减少基质干扰。在定性定量方法上,传统的ECD、FPD是通过化合物的保留时间定性,容易造成假阳性;MSD通过保留时间,标准谱库检索,碎片离子丰度比进行定性,从而有效地降低了假阳性[5]。在定量方面,MSD中的选择离子监测(SIM)方式可以在一定程度上提高化合物的信噪比(表1)。
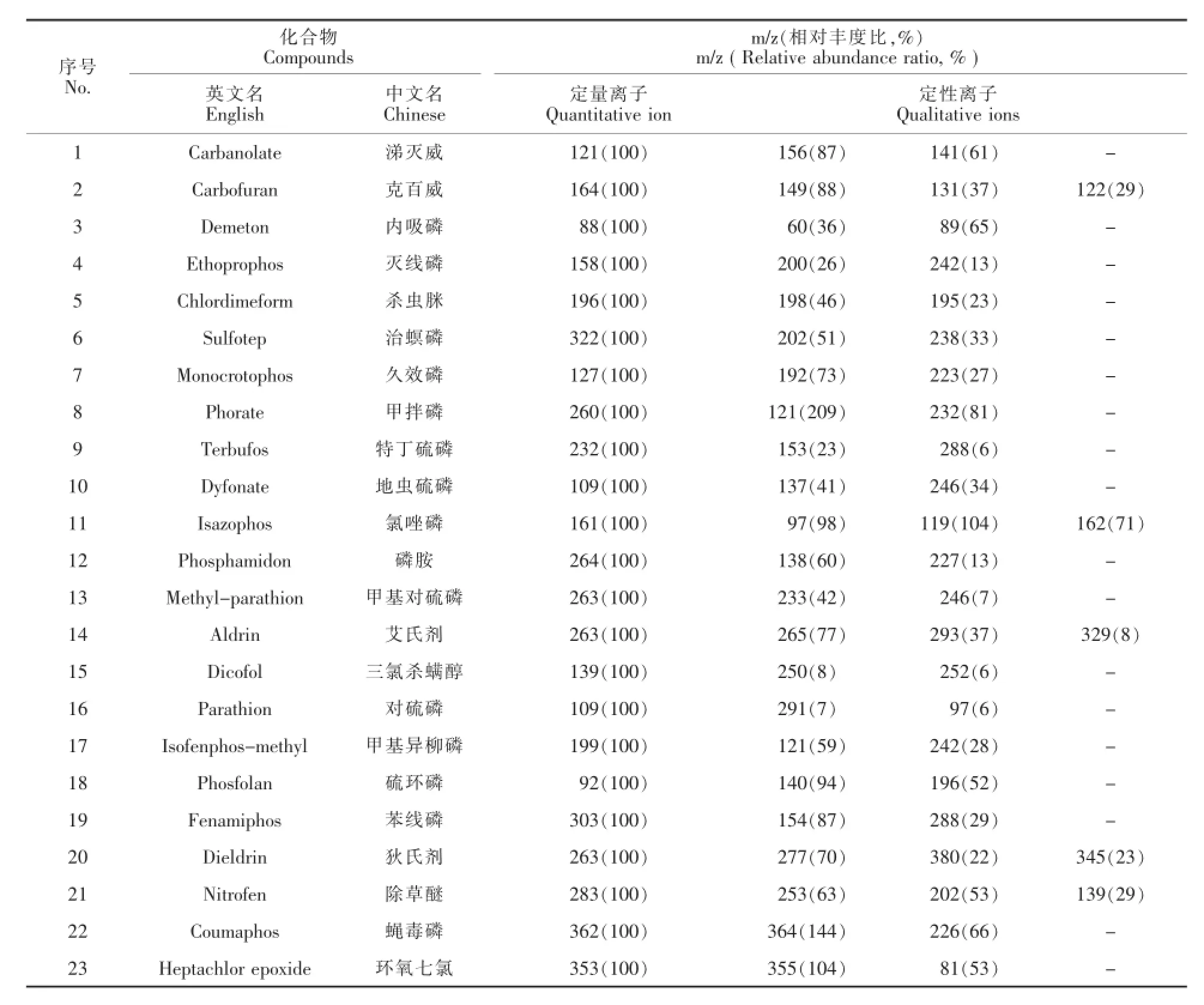
表 1 茶叶中22种农药和环氧七氯(内标)的定量离子、定性离子及丰度比Table 1 SIM parameters for GC-MS analysis of 22 pesticides
2.3方法评价
2.3.1线性方程
对基质中22种农药的0.05、0.10、0.20、0.50、1.00 μg/mL浓度进行测定,以化合物与内标物的响应峰面积比作为纵坐标,浓度作为横坐标绘制标准工作曲线 (表2),结果表明,22种农药在0.05~1.00 μg/mL的浓度范围内,其工作曲线的相关系数均大于0.99,表明该定量方法适合在该浓度范围内对样品中该22种农药的含量进行测定。
2.3.2仪器最小检出量和方法检出限
根据信噪比S/N=3计算,仪器对22种农药的最小检出量(Limit of Detection,LOD)为0.2~10.5 pg。根据信噪比S/N=10计算,该检测方法对茶叶中22种农药的检出限 (Limit of Quantification,LOQ)为0.001~0.031 mg/kg(表2)。

表2 22种农药的保留时间、标准工作曲线、最小检出量和定量限Table 2 Retention times,linear equations,correlation coefficients,detection limits of 22 pesticides
2.3.3方法准确度和精密度
在1.00、0.25、0.025 mg/kg浓度添加水平下,22种农药的回收率见表3。由表3可知,在3个不同添加浓度下,22种农药的回收率在65%~120%之间,相对标准偏差小于10.0%,该结果符合实际检测工作中对于准确度和精密度的要求(图3、4)。
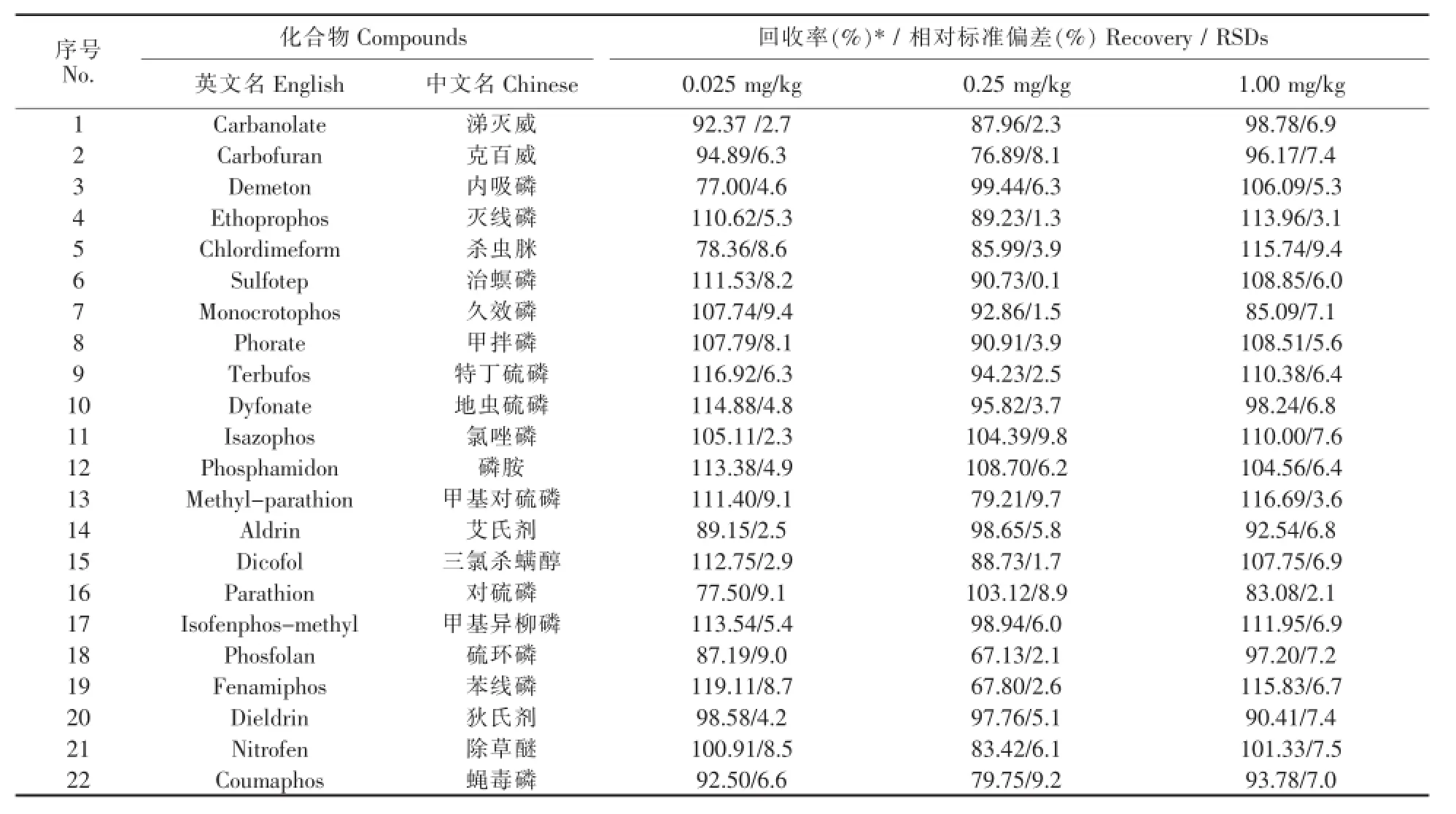
表3 22种农药在3个不同添加浓度下的回收率和相对标准偏差Table 3 Recoveries and relative standard deviations(RSDs)of 22 pesticides in tea
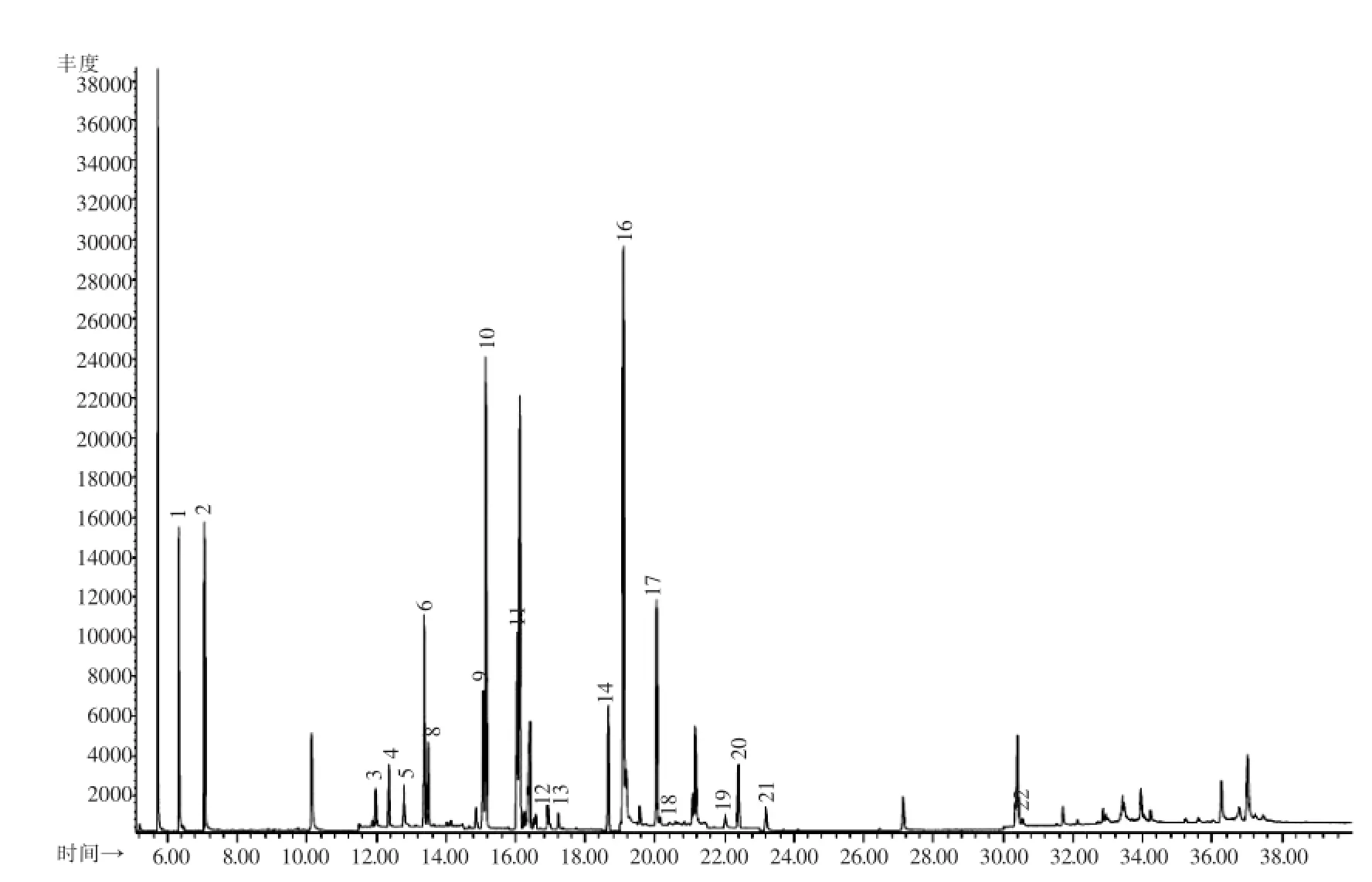
图2 茶叶基质中22种农药标准溶液的SIM谱图(0.环氧七氯;1.涕灭威;2.克百威;3.内吸磷;4.灭线磷;5.杀虫脒;6.治螟磷;7.久效磷;8.甲拌磷;9.特丁硫磷;10.地虫硫磷;11.氯唑磷;12.磷胺;13.甲基对硫磷;14.艾氏剂;15.三氯杀螨醇;16.对硫磷;17.甲基异柳磷;18.硫环磷;19.苯线磷;20.狄氏剂;21.除草醚;22.蝇毒磷)Fig.2 GC-MS SIM chromatogram of mixed pesticide standard solution(0.Heptachlor epoxide;1.Carbanolate;2.Carbofuran;3.Demeton;4.Ethoprophos;5.Chlordimeform;6.Sulfotep;7.Monocrotophos;8. Phorate;9.Terbufos;10.Dyfonate;11.Isazophos;12.Phosphamidon;13.Methyl-parathion;14.Aldrin;15.Dicofol;16.Parathion;17. Isofenphos-methyl;18.Phosfolan;19.Fenamiphos;20.Dieldrin;21.Nitrofen;22.Coumaphos)
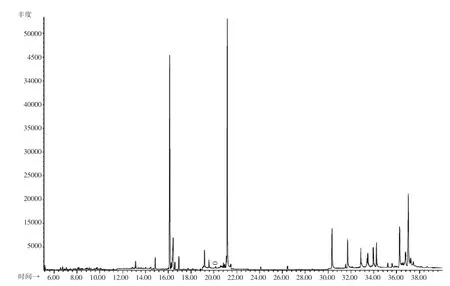
图3 茶叶基质空白对照SIM谱图(0.环氧七氯)Fig.3 GC-MS SIM chromatogram of blank tea sample(0.Heptachlor epoxide)
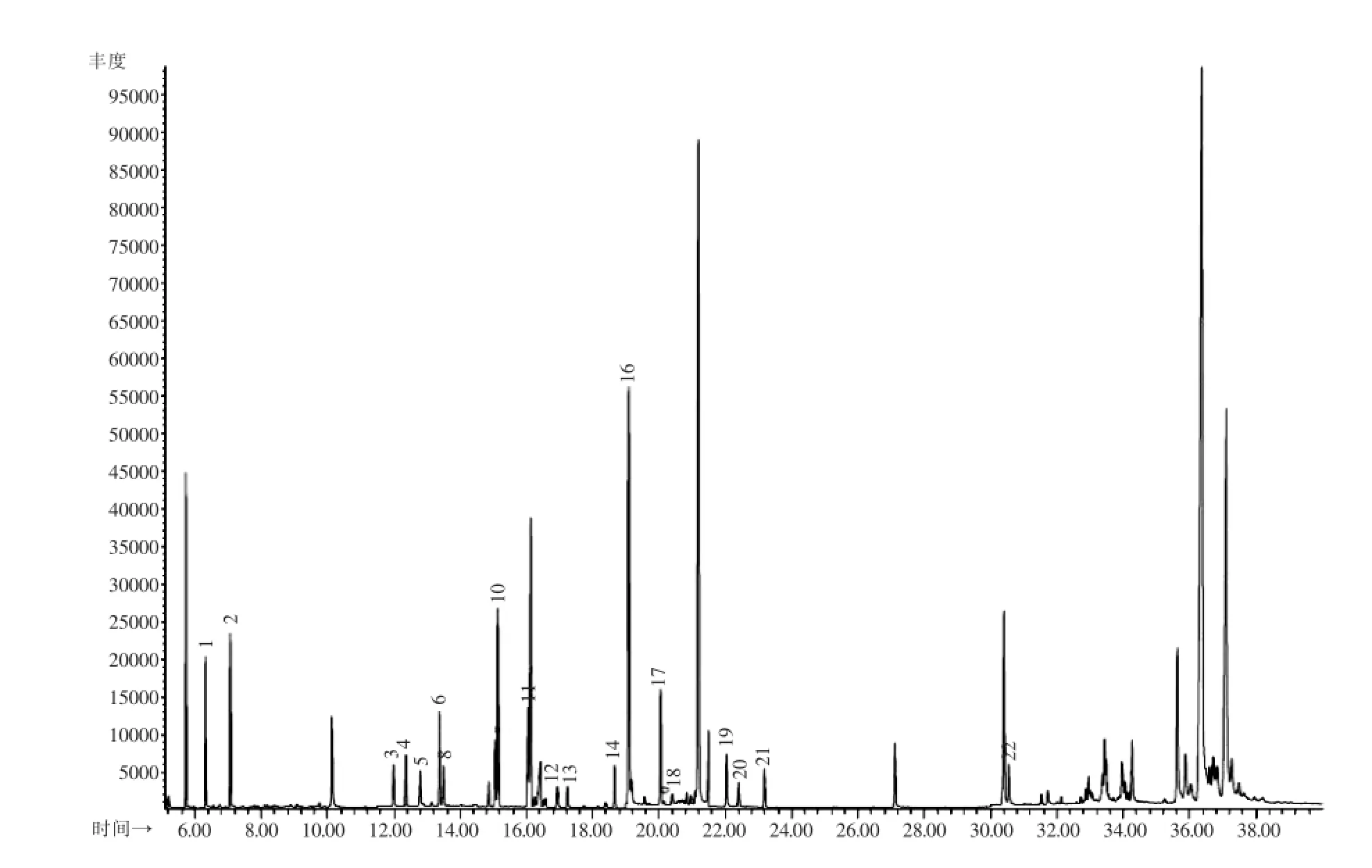
图4 22种农药添加回收试验SIM谱图(0.环氧七氯;1.涕灭威;2.克百威;3.内吸磷;4.灭线磷;5.杀虫脒;6.治螟磷;7.久效磷;8.甲拌磷;9.特丁硫磷;10.地虫硫磷;11.氯唑磷;12.磷胺;13.甲基对硫磷;14.艾氏剂;15.三氯杀螨醇;16.对硫磷;17.甲基异柳磷;18.硫环磷;19.苯线磷;20.狄氏剂;21.除草醚;22.蝇毒磷)Fig.4 GC-MS SIM chromatogram of tea sample spiked with 22 pesticides(0.Heptachlor epoxide;1.Carbanolate;2.Carbofuran;3.Demeton;4.Ethoprophos;5.Chlordimeform;6.Sulfotep;7.Monocrotophos;8. Phorate;9.Terbufos;10.Dyfonate;11.Isazophos;12.Phosphamidon;13.Methyl-parathion;14.Aldrin;15.Dicofol;16.Parathion;17. Isofenphos-methyl;18.Phosfolan;19.Fenamiphos;20.Dieldrin;21.Nitrofen;22.Coumaphos)
3 结论
该方法采用固相萃取净化技术,气相色谱-质谱分析手段,建立了快速、准确、有效地检测茶叶中的有机磷、有机氯和氨基甲酸酯类禁用农药。采用乙腈作为提取溶剂,既能有效提取各种农药,又有效地降低了基质干扰;净化过程中采用的石墨化碳-氨基混合小柱可以吸收大部分色素、糖类物质而不保留目标化合物,有效的降低了基质干扰,提高了方法的准确度和精密度。采用加标回收率试验表明,本方法定量限、回收率和相对标准偏差均满足茶叶中22种禁用农药日常检测要求。
参考文献:
[1]刘亮,孙艳萍,周蔚.我国农药禁限用政策实施情况及建议[J].农药科学与管理,2013,34(7):1-4.
[2]姚晗珺,刘欣,董国堃.两岸禁用农药比较研究[J].农药科学与管理,2011,32(7):7-10.
[3]汪云刚.茶园禁用农药简介[J].云南热作科技,2011,24(1):41.
[4]国家卫生和计划生育委员会,农业部.GB 2763-2014食品中农药最大残留限量[S].北京:中国标准出版社,2014.
[5]罗逢健,陈宗懋,汤富彬.固相萃取和气相色谱-质谱法测定茶叶中34种农药残留[J].农药,2010,49(5):363-366.
[6]王兴宁,蔡秋,朱明.固相萃取净化-气相色谱/串联质谱法测定茶叶中54种农药残留量[J].分析试验室,2011,30(11):110-116.
[7]邓美林,陈俊赤,段云鹏.凝胶色谱-气相色谱-串联质谱法筛查茶叶中27种禁用农药[J].分析实验室,2014,33(3):351-355.
[8]李军明,钟读波,王亚琴.在线凝胶渗透色谱-气相色谱/质谱法检测茶叶中的153种农药残留[J].色谱,2010,28(9):840-848.
中图分类号:S571.1;F767.2
文献标识码:A
文章编号:2095-0306(2015)01-0011-07
收稿日期:2014-09-11
作者简介:陆小磊(1984-),男,浙江瑞安人,工程师,主要从事茶叶质量安全检测工作。
Determination of 22 Banned Pesticides Residues in Tea by Solid Phase Extraction and Gas Chromatography Mass Spectrometry
LU Xiao-lei1,JIANG Mei-rong2,YE Mei-jun1,ZHOU Wei-long1
(1.Hangzhou Tea Research Institute,CHINA COOP,China National Center of Quality Supervision and Inspection of Tea,
Hangzhou 310016,China;2.Zhejiang Economic and Trade Polytechnic,Hangzhou 310018,China)
Abstract:A method was developed for determination of 22 banned pesticides residues in tea by gas chromatography mass spectrometry(GC-MS).The sample was homogenized by acetonitrile and cleaned-up by Carb-NH2SPE column,finally determinated by GC-MS coupled with DB-5 column.The Results indicated that the correlation coefficients(R)of standard working curve in tea matrix(0.05~1.00 μg/mL)were above 0.99.The recoveries of 22 pesticides were between 65%and 120%at spiked levels of 1.00,0.25 and 0.025 mg/kg,with relative standard deviations(RSDs)below 10%.The limits of quantitation(LOQ)were 0.001~0.031 mg/kg.The present method is rapid,stable and precise,and suitable for the determination of banned pesticides in tea.
Key words:Tea;Banned pesticides;Residues;Solid phase extraction;Gas chromatography mass spectrometry
猜你喜欢
化工管理(2022年13期)2022-12-02
茶叶通讯(2022年2期)2022-11-15
现代仪器与医疗(2021年2期)2021-07-21
创造(2020年5期)2020-09-10
农药科学与管理(2019年12期)2019-05-20
快乐语文(2018年36期)2018-03-12
分析化学(2016年7期)2016-12-08
肉类研究(2015年1期)2015-04-08
智慧与创想(2013年10期)2013-11-28
现代农业科技(2009年19期)2009-03-20
