
遗传性转甲状腺素蛋白淀粉样变性心肌病的临床特点
2015-08-10 09:37田庄李剑吴炜陈未张抒扬方全
中国介入心脏病学杂志 2015年5期
田庄 李剑 吴炜 陈未 张抒扬 方全
淀粉样变性是不可溶性淀粉样物质沉积于器官或组织的细胞外区,导致其功能障碍的一组疾病,其中心脏是淀粉样变性常累及的器官。心脏内沉积的淀粉样物质可以有多种,常见的有免疫球蛋白轻链、转甲状腺素蛋白(transthyretin,TTR)和心房钠尿肽[1-2]。不同淀粉样物质沉积导致的心脏受累和心脏外的表现有所不同。TTR 由肝细胞合成,正常状态下为四聚体,参与甲状腺素和维生素A 的转运,形成淀粉样纤维沉积后会导致2 种淀粉样变,一种是野生型基因产生的TTR 形成的系统性老年淀粉样变,多见于70 岁以上男性;另一种是基因突变导致TTR 构象发生改变、沉积导致的遗传性TTR 型淀粉样变(transthyretin amyloidosis,ATTR)[3-7]。本文总结北京协和医院收治的遗传性ATTR 心肌病患者的临床特点,以提高临床对该病的认识和诊治。
1 对象与方法
1.1 研究对象
检索1999 年至2014 年于北京协和医院住院诊断为系统性淀粉样变性的患者,依据病理检查和基因检测结果,共纳入13 例遗传性ATTR 心肌病变患者;选取符合标准的免疫球蛋白轻链沉积所致原发性淀粉样变(light-chain amyloidosis,AL)累及心脏的患者58 例作为对照组进行比较。
1.1.1 入选标准 (1)所有患者淀粉样变诊断明确,即均经组织(齿龈、皮肤、骨髓、腹部脂肪、胃肠黏膜以及心肌组织中的1 种或2 种以上组织)病理检查刚果红染色阳性;(2)淀粉样变心肌病诊断依据:①心内膜活检刚果红染色阳性;②如果未行心内膜活检,其他组织活检显示刚果红染色阳性,同时超声心动图提示有浸润性心肌病表现(室壁增厚>12 mm、心肌回声增强,特别是颗粒样回声增强)[8];(3)经过免疫组织化学染色或蛋白组学方法明确淀粉样物质为轻链型诊断为AL;经以上方法明确为TTR 物质,再经基因检测有TTR 基因突变的为遗传型ATTR;(4)所有患者具有完整病史资料记录、实验室检查、心电图和超声心动图检查结果。
1.1.2 排除标准 (1)临床可疑、但无病理学明确淀粉样变诊断。(2)淀粉样变分型不明确。(3)继发于多发性骨髓瘤或其他浆细胞疾病的淀粉样变。(4)无心脏受累证据。(5)临床资料不完善患者。
1.2 研究方法
观察患者临床资料,包括(1)年龄、性别、病史长短、临床主要表现。(2)受累器官情况,包括周围神经病变、自主神经病变、肾病变、消化系统病变及皮肤出血等;周围神经病变定义为患者出现周围神经功能障碍相关的症状和(或)体征;自主神经病变定义为出现少汗、肠道功能异常(腹泻和便秘交替)、体位性低血压、勃起异常及尿潴留等;肾受累指24 h 尿蛋白定量大于500 mg;消化系统受累指有消化道组织病理显示淀粉样物质沉积;肝受累是以碱性磷酸酶(ALP)升高至正常值上限的1.5 倍为标准[8]。(3)实验室检查包括血肌酸酐、ALP 和B 型脑钠肽(BNP)以及24 h 尿蛋白定量。(4)超声心动图和心电图结果。超声心动图包括胸骨旁长轴切面测量的左心房内径、左心室舒张末内径、左心室射血分数(LVEF)、室间隔和左心室后壁厚度;根据三尖瓣反流速度计算的肺动脉收缩压力。心电图结果包括心律、是否有病理性Q 波、导联低电压以及传导阻滞(包括房室传导阻滞和束支传导阻滞)。
1.3 统计学分析
采用SPSS 19.0 软件进行数据处理。计量资料以 珋x±s 或中位数(四分位间距)表示,符合正态分布的数据采用t 检验,反之采用Mann-Whitney U 检验;计数资料以例数(百分比)表示,采用卡方检验或精确概率检验。BNP 不符合正态分布,将其进行lg 转换后符合正态分布,两组之间比较采用t 检验。以P <0.05 为差异有统计学意义。
2 结果
2.1 基线资料
13 例遗传性ATTR 心肌病患者中,男10 例(76.9%),平均年龄26.0 ~62.0(47.0 ±11.0)岁。临床主要表现为慢性腹泻4 例(30.8%),胸闷、气短3 例(23.1%),周围神经病变(四肢麻木、肌肉萎缩等)3 例(23.1%,其中2 例有轻微慢性腹泻),头晕或肢体障碍2 例(15.4%)及视物不清1 例(7.7%)。因以上症状分别就诊于消化科、心内科、神经内科和眼科。病程17.0(14.0,27.0)个月,起病至发现心肌病变时间为13.0(11.0,21.0)个月。9 例(69.2%)患者有类似临床表现家族史[周围神经病变和(或)心肌肥厚]。与AL 患者相比,ATTR患者更年轻[(47.0 ±11.0)岁比(57.0 ±10.0)岁,P<0.01]、病程[17.0(14.0,27.0)月比11.0(6.0,13.0)月,P <0.01]及起病至发现心肌病变[13.0(11.0,21.0)月比2.5(1.0,5.0)月,P <0.01]时间更长,差异均有统计学意义(表1)。
2.2 心肌病变情况
遗传性ATTR 心肌病患者的临床表现主要为活动耐力减低4 例(30.8%)和下肢水肿1 例(7.7%),8 例(61.5%)患者并无心脏受累的明显临床表现,经超声心动图和心电图检查发现有心肌病变;NAYH 心功能分级Ⅰ~Ⅱ级患者11 例(84.6%),Ⅲ级患者2例(15.4%);与AL型心肌病患者相比(Ⅲ~Ⅳ级),遗传性ATTR 心肌病患者的临床心功能情况更好(15.4%比50.0%,P <0.05);同时ATTR 组的血浆BNP 值也显著低于AL 组[126.0(73.0,330.0)ng/L 比 664.0 (385.0,1568.0)ng/L,P <0.01,表1)]。
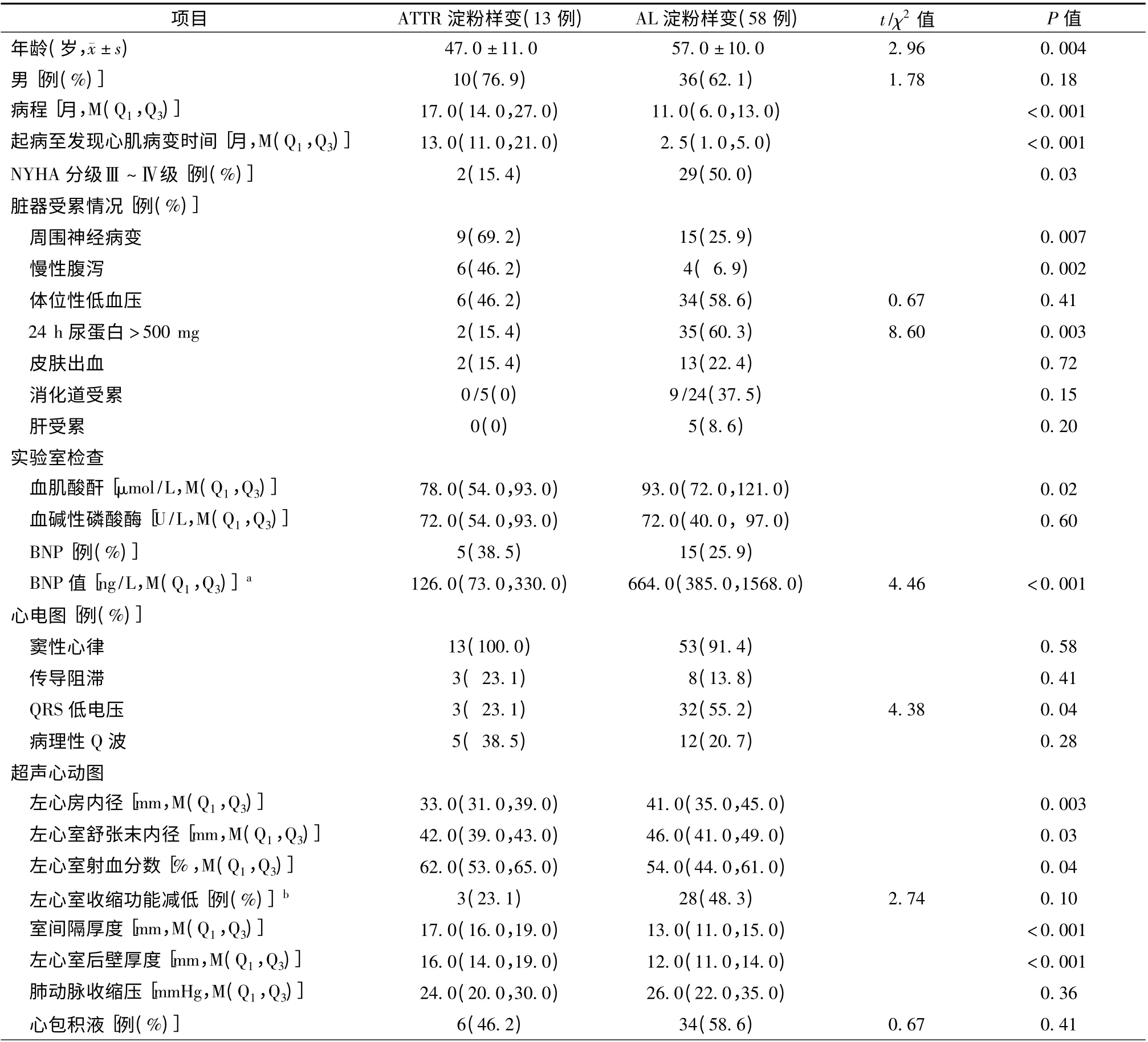
表1 ATTR 淀粉样变心肌病与AL 淀粉样变心肌病患者临床资料对比
心电图检查显示,遗传性ATTR 心肌病患者均为窦性心律,3 例(23.1%)出现传导阻滞,包括房室传导阻滞1 例(7.7%)和束支传导阻滞2 例(15.4%),与AL 心肌病患者比较,差异均无统计学意义(P >0.05);遗传性ATTR 患者3 例(23.1%)显示QRS 波低电压及5 例(38.5%)显示有病理性Q 波,QRS 低电压发生率显著低于AL 患者(23.1%比55.2%,P=0.04)。超声心动图示,遗传性ATTR心肌病变患者主要为心室壁均匀增厚,室间隔厚17.0(16.0,19.0)mm,左心室后壁16.0(14.0,19.0)mm;左心房内径33.0(31.0,39.0)mm,左心室无增大,舒张末期内径42.0(39.0,43.0)mm,大多数患者收缩功能正常,仅3 例(23.1%)患者LVEF <55.2%。与AL 患者相比,遗传性ATTR 心肌病患者左心房内径(P <0.01)和左心室内径(P <0.05)均较小,左心室室壁肥厚更为明显(P <0.01)。此外淀粉样心肌病患者容易合并心包积液,ATTR 型6 例(46.2%),AL 型34 例(58.6%)。虽然超声心动图显示ATTR 组患者存在有左心室室壁明显增厚,但未见患者的心电图提示有QRS 波高电压(表1)。
2.3 实验室检查
ATTR 心 肌 病 患 者 血 肌 酸 酐 水 平78.0(54.0,93.0)μmol/L,明 显 低 于AL 型 患 者93.0(72.0,121.0)μmol/L(P <0.05);24 h 尿蛋白定量0.67(0.3,1.0)g,较AL 患者1.24(0.5,4.6)g低,但差异无统计学意义(P >0.05);24 h 尿蛋白定量>500 mg的患者2 例(15.4%),显著少于AL 患者35 例(60.3%),差异有统计学意义(P <0.05);
2.4 心脏外表现
遗传性ATTR 心肌病患者除心脏受累外,还有心脏外受累。与AL 患者相比,遗传性ATTR 心肌病患者周围神经病变(69.2%比25.9%,P <0.01)和慢性腹泻(46.2%比6.9%,P <0.01)更为多见,肾受累更为少见(15.4%比60.3%,P <0.01),差异均有统计学意义。ATTR 组患者未出现肝受累(ALP均在正常范围内),而AL 患者中有5 例(8.6%)出现ALP 的升高。5 例(38.5%)遗传性ATTR 心肌病患者进行胃肠黏膜活检和刚果红染色,均未发现有淀粉样物质沉积,AL 患者中有24 例进行活检,9 例(37.5%)阳性。ATTR 与AL 患者体位性低血压和皮肤出血发生率比较,差异均无统计学意义(P >0.05,表1)。
3 讨论
遗传性ATTR 淀粉样变是常染色体显性遗传,该基因位于18 号染色体,发生突变后生成异常TTR蛋白,容易从四聚体解离为单体,形成β 折叠层纤维并沉积在多个组织器官从而导致系统性淀粉样变[1]。受到突变基因、地域、年龄和性别等多种因素影响,遗传性ATTR 临床表型多样。以周围神经病变为主要表现,被称为家族性淀粉样变性多发神经病变;也可以导致心脏病变,表现为活动耐力减低和右心衰竭,或者心电图和超声心动图发现有异常;有些可以表现为慢性腹泻或玻璃体浑浊,甚至可以无任何明显临床表现[1-7]。因此,遗传性ATTR 患者可就诊于心内科以外的多个科室,如消化科、神经内科或者眼科。
遗传性ATTR 为常染色体显性遗传疾病,患者多存在周围神经疾病或心肌疾病家族史。本研究中9 例患者直系亲属中有该病的相关症状,尽管未进行基因测定,考虑到为同一基因突变所致可能性大。另外4 例患者没有明确家族史,考虑到遗传性ATTR 并不一定累及心脏或心脏受累而症状比较隐匿[9],由于并未进行家系基因测定和心脏方面的检查,并不能完全除外家族其他成员患病的可能。本研究与既往研究结果相一致,显示遗传性ATTR 患者起病年龄较AL 型患者更早,特别是合并有周围神经病变患者[2]。考虑是ATTR 为遗传性疾病的缘故,虽然起病较早,但疾病进展速度较AL 型慢,病程较长。
与AL 相似,遗传性ATTR 也是系统性疾病,累及多个脏器。心脏的各个部分包括心室、房间隔、传导系统、瓣膜以及冠状动脉均可受累,最常见表现为心肌病变和传导阻滞[2-7,10-11]。由于淀粉样物质沉积在心室壁中,心肌病变主要表现为左心室或左、右心室肥厚。与AL 患者相比,遗传性ATTR 心肌病患者室壁肥厚更明显,而左心室收缩功能和临床心功能状况较好,BNP 数值也明显较低[2,4,11]。另外淀粉样变心肌病的心电图特征性表现是缺乏与室壁肥厚相对应的QRS 波高电压,甚至会出现低电压,这是淀粉样心肌病与肥厚型心肌病和血流动力学改变导致的左心室肥厚(如高血压、主动脉瓣狭窄等)鉴别要点[2-5]。本研究显示,遗传性ATTR 患者主要是缺乏QRS 高电压表现,低电压发生率较AL 心肌病低。而有研究指出,无心电图导联低电压的左心室肥厚并不能排出淀粉样变性,特别是TTR 型淀粉样心肌病的可能;遗传性ATTR 与AL 心肌病之间超声心动图和心电图的差异,考虑与单克隆轻链沉积速度较快以及其对心肌细胞的毒性等因素相关[2,12]。此外,遗传性ATTR 心肌病患者的心电图表现传导异常也比较多见,特别是束支传导阻滞。
如前所述,除心脏外,遗传性ATTR 与AL 均可累及全身多个系统,包括肾、肝、胃肠道、周围神经和自主神经等。与AL 患者相比,遗传性ATTR 更多合并周围神经病变和慢性腹泻,较少出现肾和肝受累[2,12]。某些基因突变可以仅表现为周围神经病变,以下肢小纤维病变起病,自远段向近段进展,以感觉异常、迟钝多见,伴或不伴疼痛;而运动异常通常出现较晚。此外自主神经也经常受累,表现为慢性腹泻为主,间断便秘以及体位性低血压。由于腹泻对患者进行肠镜检查,往往不能发现淀粉样物质沉积,考虑并非直接侵犯肠道所致。遗传性ATTR心肌病患者仅有少量蛋白尿,血肌酸酐基本正常或者仅轻度升高,ALP 多正常[1];而AL 患者的肾受累比较明显,较多出现大量蛋白尿以及血肌酸酐的升高;肝脏受累以及肠道淀粉样物质的沉积也相对较多。
本研究显示,遗传性ATTR 是由于TTR 基因突变产生异常蛋白、形成淀粉样纤维沉积所致。心脏受累的主要表现为室壁明显增厚而心电图并无相应的QRS 波高电压,左心室通常未见增大而收缩功能正常。除心脏受累外,多合并有周围神经病和自主神经受累(腹泻、体位性低血压)。由于是常染色体显性遗传,该病有家族聚集性。
本研究为回顾性研究,ATTR 患者数量较少,且并非每例患者均详细筛查是否存在器官受累以及病变程度(如胃肠道活检、血BNP 和24 h 尿蛋白测定);未对患者进行家系基因检测和心脏检查;超声心动图检查亦不完善,缺乏右心室室壁厚度、瓣膜受累情况以及左心室舒张功能的判定,还需进一步临床研究进行证实。
[1] Dungu JN,Anderson LJ,Whelan CJ,et al. Cardiac transthyretin amyloidosis. Heart,2012,98:1546-1554.
[2] Rapezzi C,Merlini G,Quarta CC,et al. Systemic cardiac amyloidosis:Disease profiles and clinical courses of the 3 main types. Circulation,2009,120:1203-1212.
[3] Sattianayagam PT,Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant.Eur Heart J,2012,33:1120-1127.
[4] Quarta CC,Solomon SD,Uraizee I,et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis.Circulation,2014,129:1840-1849.
[5] Rapezzi C,Quarta CC,Obici L,et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J,2013,34:520-528.
[6] Arruda-Olson AM,Zeldenrust SR, Dispenzieri A, et al.Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid,2013,20:263-268.
[7] Rapezzi C,Longhi S,Milandri A,et al. Cardiac involvement in hereditary-transthyretin related amyloidosis. Amyloid,2012,19(Suppl 1):16-21.
[8] Gertz MA,Comenzo R,Falk RH,et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis,Tours,France,18-22 April 2004. Am J Hematol,2005,79:319-328.
[9] Hellman U,Alarcon F,Lundgren HE,et al. Heterogeneity of penetrance in familial amyloid polyneuropathy,ATTR Val30Met,in the Swedish population. Amyloid,2008,15:181-186.
[10] 许芷绮,盛琴慧.碎裂QRS 波的研究进展及临床应用.中国介入心脏病学杂志,2013,21:192-194.
[11] Connors LH,Prokaeva T,Lim A,et al. Cardiac amyloidosis in African Americans:comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J,2009,158:607-614.
[12] Brenner DA,Jain M,Pimentel DR,et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res,2004,94:1008-1010.
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2022年4期)2022-05-23
中国典型病例大全(2022年10期)2022-05-10
中国听力语言康复科学杂志(2021年6期)2021-12-21
保健医苑(2020年10期)2020-11-11
保健文汇(2020年7期)2020-08-21
保健与生活(2020年13期)2020-07-24
中国临床医学影像杂志(2019年1期)2019-04-25
