
Catalytic Hydrogenation of Methanol-Containing Effluent from Epoxidation of Propylene
2015-06-22 14:38
中国炼油与石油化工 2015年3期
(SINOPEC Luoyang Petrochemical Complex, Luoyang 471012)
Catalytic Hydrogenation of Methanol-Containing Effluent from Epoxidation of Propylene
Cheng Ke
(SINOPEC Luoyang Petrochemical Complex, Luoyang 471012)
This paper describes the hydrogenation of impurities in the methanol-containing effluent from the propylene epoxidation process with hydrogen peroxide. The effects of reaction temperature, pressure, weight hourly space velocity (WHSV) and H2/methanol ratio on the concentration of various impurities in methanol solvent were investigated. It was found out that the aldehyde, hydrogen peroxide and nitro compounds in the methanol solvent could be completely hydrogenated over the Ni catalyst under proper reaction conditions. 90% of acetone and up to 50% of acetals (ketals) existing in the methanol solvent could be hydrogenated. No significant change was observed for the rest of the impurities that were present in the methanol solvent (i. e., 1-methoxy-2-propanol, 2-methoxy-1-propanol and 1,2-propanediol). The H2O2decomposition reaction was enhanced using Ni catalyst, through the formation of NioOH, but no oxygen was found in the off-gas of hydrogenation reaction since NioH could react on NioOH formed via dissociative adsorption of hydrogen peroxide, or on NioO formed via adsorption of oxygen.
propylene; epoxidation; methanol; hydrogenation
1 Introduction
Propylene oxide (PO) is a major propylene derivative amounting to more than 6 million tons per annum worldwide. World propylene oxide consumption in 2005 was 6.213 million tons and reached 6.753 million tons in 2008[1-2]. During the 2009-2010 period, the worldwide consumption of propylene oxide fell about 3% to 4% owing to the full impact of global recession. This decline would have been worse if it was not bolstered by Asian demand and the needs of its fastest-growing user— China. But the worldwide consumption of propylene oxide had grown strongly after 2010 and reached over 7.7 million tons in 2012. By 2015, the world demand for propylene oxide is expected to reach 9.72 million tons.
Propylene oxide is currently produced by three different types of commercial processes: the chlorohydrin process (CHPO)[3-4], the propylene co-oxidation process (CHP, PO/SM and PO/TBA)[5-7]and the direct propylene oxidation process. Because of the negative environmental impact of the chlorohydrin process[8]and the depressed co-products market of the propylene co-oxidation process[9-10], most recently built propylene oxide plants have chosen the direct oxidation process, using the HPPO (Hydrogen Peroxide to Propylene Oxide) technology[11-12]. The HPPO process only produces propylene oxide and water, without other products associated with the traditional processes. This process also avoids the generation of such by-products as propylene dichloride, MTBE and styrene monomer. In addition, this process has been proven to be more cost-effective and environmentally friendly[13-15].
In the HPPO process, methanol is typically used as the solvent to improve the solubility of propylene in the reaction mixture[16]. After propylene recovery and propylene oxide separation, the solvent methanol must be recovered from the waste water and be recycled in the epoxidation process. Besides water, the methanol-containing effluent from the product separation unit also contains various impurities such as acetaldehyde, propylene glycol, propylene glycol monomethyl ether, acetone and ethylene glycol dimethyl ether, and it is difficult to get relatively high purity methanol simply by means of the distillation process. Meanwhile, in spite of the high conversion of hydrogen peroxide over titanium catalysts in the epoxidation process[17-20], an 100% conversion of hydrogen peroxide cannot be achieved in the epoxidation reactor. Small amountsof residual hydrogen peroxide at the exit of the epoxidation reactors cannot be introduced into the methanol purification steps, because the decomposition of hydrogen peroxide will generate oxygen, causing serious safety problems[21].
An attractive alternative process involves the hydrogenation of the methanol-containing effluent, and a series of successive reactions which convert the impurities to some other species with high boiling point and convert residual hydrogen peroxide into water without the formation of flammable gases. However, previous investigations on the hydrogenation of the effluent are limited and scatted. Philip Landon[22], et al. described the direct synthesis of hydrogen peroxide from H2and O2using a range of supported noble metal catalysts and discussed the decomposition of hydrogen peroxide in the presence of hydrogen. Brieva[23], et al. studied the selective catalytic hydrogenation of a solution that simulates the epoxidation reactor exit stream of HPPO process and investigated the effect of the reaction temperature, catalyst amount and hydrogen partial pressure on catalyst performance. Thomas Haas[24], et al. proposed a catalyst comprising 2% Ru on activated carbon for the hydrogenation of methanol-containing effluent and the result showed that carbonyl compounds like acetaldehyde and formaldehyde could be selectively removed. Other researchers focused their interests[25-28]on the hydrogen peroxide decomposition over different catalysts in the presence or absence of hydrogen.
The purpose of this work is to study the selective catalytic hydrogenation of impurities in the effluent of the epoxidation process. In this work, the supported nickel catalyst was prepared and evaluated in this reaction in order to find an effective method to eliminate impurities, which were not referred to in the technical literature. Furthermore, this paper will focus on the effects of reaction temperature, pressure, WHSV and H2/effluent ratio on the concentration of various impurities in the solution.
2 Experimental
2.1 Apparatus
The hydrogenation of the solution was carried out in a fixed-bed microreactor. In the hydrogenation experiments, the temperature around the reactor tube was monitored and controlled using a PID controller. The reactor filled with catalyst (10 mL in volume) was connected to a separator which was immerged in the water bath. The reaction pressure was controlled by the valve in the gas outlet pipeline of the separator. Prior to the reaction, the reactor was pressurized to 1 MPa, and flushed with N2and H2at desired rates. Then, the pressure was increased to the specified reaction pressure, and the temperature was increased to the required reaction temperature. The effluent from the reactor was introduced to a separator where the gas (mainly hydrogen) passed through the pressure valve and the liquid was collected separately. The concentration of the organic compounds was determined by GC using a Varian CP-3800 instrument fitted with a Varian capillary column. The conversion rate of impurities in the solution was calculated according to the following equation:

where Sinand Soutrepresent the concentration of impurities in the solution entering (in) and leaving (out) the reactor.
2.2 Feedstock
The reaction mixture was prepared to simulate the effluent in a propylene epoxidation process after elimination of propylene and propylene oxide. Table 1 summarizesthe composition of the effluent. Further analyses showed that the mass fraction of sulfur (S) in the solution was as low as 4 μg/g, however, the mass fraction of nitrogen (N) reached as high as 188 μg/g. Nitro compounds such as nitromethane and nitroethane were formed from auxiliary agents added into the epoxidation process. Because of the small amount of hydrogen peroxide remaining in the solution, the methanol-water solution had a pH value of less than 7.

Table 1 Composition of methanol water solution
2.3 Catalyst preparation and characterization
Alumina-supported nickel catalyst (Ni/Al2O3) was employed in the experiments. All supported nickel catalysts were prepared by the one-step incipient impregnation method[29]. The supports were saturated in normal aqueous solutions of Ni(NO3)2·6H2O at 40 ℃ using a thermostatic bath, and then dried at 90 ℃ in an oven for 16 hours, calcined in air at 500 ℃ in a furnace for 6 hours, crushed and sieved to a particle size of between 0.180 mm and 0.154 mm. Prior to the activity evaluation, the catalyst samples were reduced at 460 ℃ for 4 hour with a mixture of hydrogen and nitrogen. All the catalyst samples were kept in a dessiccator prior to being used in the experiments. Physico-chemical properties of the catalyst are presented in Table 2.

Table 2 Physico-chemical properties of Ni/γ-Al2O3catalyst
3 Results and Discussion
The Ni/Al2O3catalyst was tested on the conversion of impurities in the solution. Its activity did not change after 2000 hours of use at different temperatures, as compared with the activity of the fresh catalyst. The result indicated that the Ni/Al2O3catalyst was appropriate to the target reactions. Experimental results have shown a strong influence of the reaction parameters, such as pressure, temperature, WHSV and H2/effluent ratio, on the conversion of impurities in the presence of H2. Table 3 shows a typical composition of solution treated by hydrogenation process at a temperature of 150 ℃ and a pressure of 4.0 MPa. The contents of hydrogen peroxide, acetaldehyde, propionaldehyde and nitro compounds were not detected; however, the mass fractions of dimethoxyethane, dimethoxypropane stayed almost unchanged.
It is clear that hydrogen peroxide was not the only compound that could be hydrogenated under these reaction conditions, since acetaldehyde and propionaldehyde could also be hydrogenated. Hydrogenation of acetaldehyde resulted in the formation of ethanol, while propionaldehyde hydrogenation would produce n-propanol. Isopropanol was formed by the partial hydrogenation of acetone. Nitro compounds could be totally converted into amine compounds, making the hydrogenated solution more alkaline. It is also found out that in the presence of hydrogen, acetal and ketal (such as dimethoxyethane and dimethoxy-propane) could also be partially hydrogenated, and alcohols and ethers would possibly be formed. No significant changes were observed in the rest of the compounds thatwere present in the solution, such as methanol, propylene glycol and propylene glycol monomethyl ether.
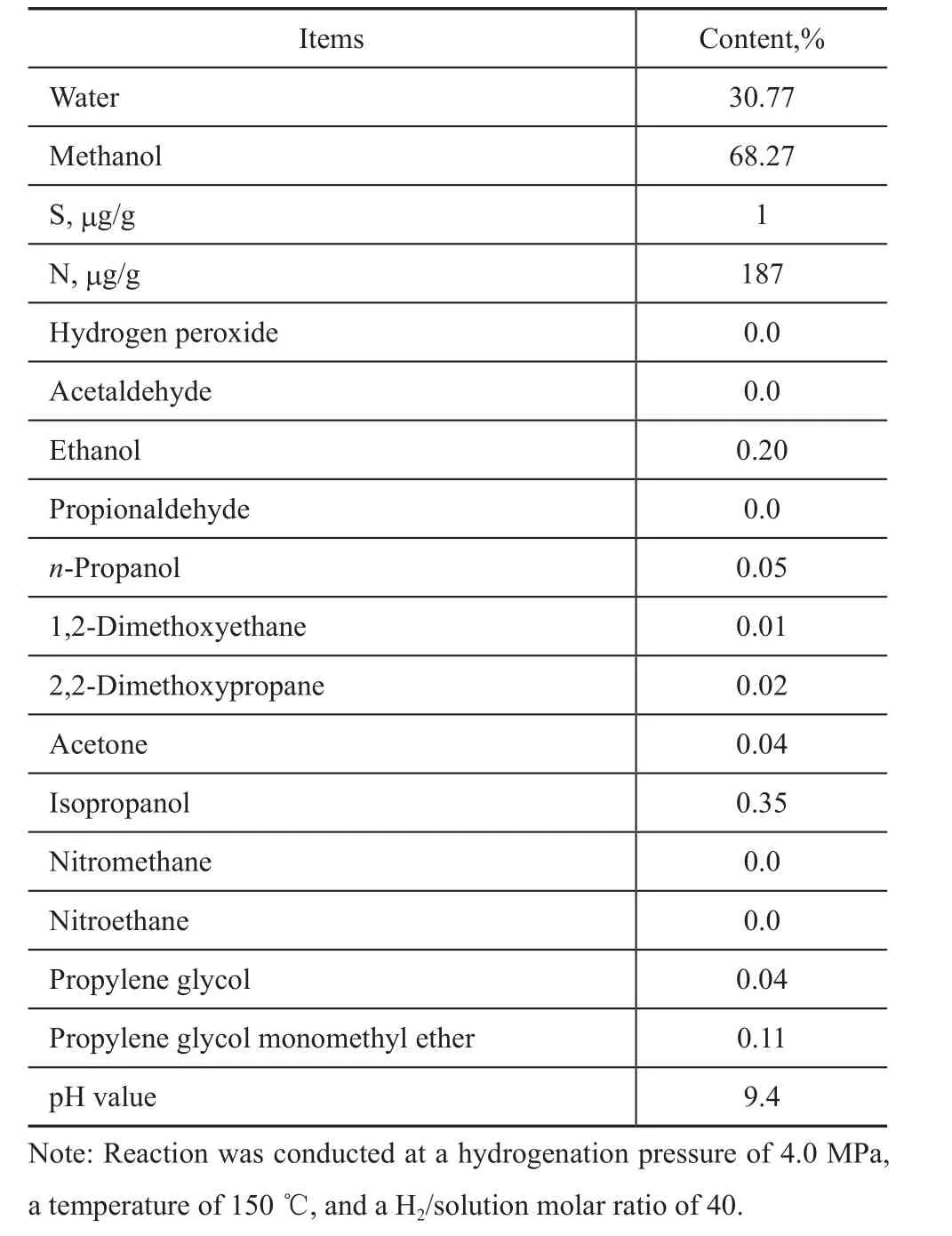
Table 3 Typical composition of the solution after hydrogenation
The hydrogenation of impurities in the methanol-water solution is thus a complex process. It is strongly influenced by the reaction and catalyst parameters, which will be discussed in the following sections.
3.1 Influence of H2/solution molar ratio
Figure 1 shows the influence of the molar ratio of H2/solution on the conversion of impurities at 4.0 MPa and 150 ℃. With the H2/solution molar ratio increasing to 10, the conversion rate of acetaldehyde, nitro compounds and hydrogen peroxide was all increased to almost 100%, while the conversion rate of acetone reached 74%. When the H2/solution ratio was increased from 20 to 40, the conversion rate of dimethoxyethane and dimethoxypropane was also increased from 30% to 50%, while the conversion rate of acetone reached 96%. The higher the molar ratio of H2/solution is, the better the removal rate of impurities would be. However, it also indicates an increased recycle rate of the hydrogen, leading to an increase in investment and operating cost. It can be seen from Figure 1 that when the H2/solution molar ratio was greater than 40, the increase in the removal of impurities gradually tapered off, indicating that an appropriate H2/solution molar ratio should be no more than 40.

Figure 1 Effect of H2/solution molar ratio on the conversion of impurities in solution
3.2 Influence of weight hourly space velocity (WHSV)
The Ni/Al2O3catalyst was also employed in the hydrogenation experiments at 4.0 MPa and 150 ℃ and a H2/solution ratio of 40. The influence of WHSV on the conversion of impurities was investigated, with the results shown in Figure 2. It was observed that when the WHSV was increased from 1.0 h-1to 6.0 h-1, the conversion rate of acetone, dimethoxyethane and dimethoxypropane decreased from 92% and 67% to 69% and 33.3%, respectively, which indicated that the reduced reaction time could result in a decreased depth of once-through conversion of impurities. The conversion of acetaldehyde, hydrogen peroxide and nitro compounds over the Ni/Al2O3catalyst still reached 100% in a range of WHSV between 1.0 h-1and 4.0 h-1, denoting that acetaldehyde, hydrogen peroxide and nitro compounds were readily susceptible to hydrogenation.

Figure 2 Effect of WHSV on the conversion of impurities in solution
It was observed that when the WHSV was increased from 1.0 h-1to 6.0 h-1, the conversion rate of acetone, dimethoxyethane and dimethoxypropane decreased from 92% and 67% to 69% and 33.3%, respectively, indicating that the reduced reaction time could result in a decreased depth of once-through conversion of impurities. The conversion of acetaldehyde, hydrogen peroxide and nitro compounds over the Ni/Al2O3catalyst still reached 100% in a range of WHSV between 1.0 h-1and 4.0 h-1, denoting that acetaldehyde, hydrogen peroxide and nitro compounds were readily susceptible to hydrogenation.
3.3 Influence of reaction pressure
The influence of reaction pressure on conversion of impurities was also examined in a pressure range of between 1.0 MPa and 5.0 MPa at a reaction temperature of 150 ℃,a WHSV of 2.0 h-1and a H2/solution ratio of 40. Above a pressure of 2.0 MPa, the conversion rate of acetaldehyde, hydrogen peroxide and nitro compounds was unchanged (almost 100%), as shown in Figure 3. At lower pressures, the efficiency of the catalyst for hydrogenation of acetone, dimethoxyethane and dimethoxypropane was reduced. With the increase of reaction pressure, the conversion rate of acetone was increased from 69% under a pressure of 2.0 MPa to 92% under a pressure of 4.0 MPa. At the same time, the conversion rate of dimethoxyethane and dimethoxypropane was increased from 17% to 50%. There was no significant change in the conversion of all the impurities when the pressure was higher than 4 MPa. Therefore, a pressure of 4.0 MPa is the appropriate reaction pressure for the hydrogenation process.

Figure 3 Influence of pressure on the conversion of impurities in solution
3.4 Influence of reaction temperature
The reaction temperature was varied from 120 ℃ to 150 ℃at a constant reaction pressure of 4.0 MPa. The dependence of the conversion of impurities in the solution on temperature is illustrated in Figure 4. It was observed that the conversion rate of acetone was increased from 77% to 95% as the reaction temperature increased from 120 ℃ to 150 ℃. At lower temperatures (below 130 ℃), the conversion rates of dimethoxyethane and dimethoxypropane were very low. The conversion rates of dimethoxyethane and dimethoxypropane increased slowly when the temperature increased to 150 ℃, then they remained basically constant with a further increase in temperature. In a temperature range between 120 ℃ to 150 ℃, the acetaldehyde and hydrogen peroxide, as well as the nitro compounds, were almost completely hydrogenated.
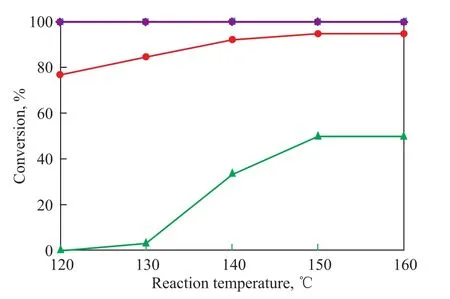
Figure 4 Effect of temperature on the conversion of impurities in solution
3.5 Hydrogenation of hydrogen peroxide
Hydrogen peroxide can be easily decomposed into water and oxygen at high temperaure even without catalysts. The decomposition reaction is highly favored thermodynamically. If its decomposition produces oxygen during hydrogenation of the solution, this oxygen product can produce serious safety problems. Therefore, the influence of hydrogen peroxide concentration on the extent of converstion was also investigated under different conditions, such as loading catalyst with hydrogen, loading catalyst without hydrogen, and loading no catalyst with no hydrogen. The hydrogenation and decomposition processes of the solution were performed at a reaction pressure of 4.0 MPa and a reaction temperature of 150 ℃. The results are shown in Figure 5.
It can be seen from Figure 5 that the decompositon of a 0.1% hydrogen peroxide solution could reach 88% even when there was neither catalyst nor hydrogen. The decomposition reached over 95% when the concentration of hydrogen peroxide in the solution was over 0.5%. When the Ni catalyst was employed in the decomposition reaction, the decompositon of hydrogen peroxide at different concentrations remained at over 98%, indicating that Ni was also active in the hydrogen peroxide decompositon process. Just like the mechanism of noble metals (Pt and Pd) that would catalyze the decomposition of hydrogenperoxide[28,31-32], the H2O2destruction over the Ni catalyst could be explained by the decomposition mechanism and properties of the Ni surface, as presented by the following equations:

Figure 5 Relationship between hydrogen peroxide concentration and its conversion rate

When Ni catalyst and hydrogen were used under the same condition, the hydrogen peroxide at different concentrations could be completely removed. Analysis of the tail gas showed that the concentration of the oxygen was 0.01%, which was almost the same as that contained in the fresh hydrogen, as shown in Table 4.
It was clear that no oxygen was formed during the hydrogenation process of the solution. The reason for such a performance could be explained by the dissociation of H2and O2into the Ni clusters, as shown below:

The first steps in the surface catalyzed hydrogen peroxide hydrogenation covered the absorption of molecular hydrogen (reaction 5) and molecular hydrogen peroxide (reaction 2). The surface bound species further reacted with each other to form the reduced Ni and water (reaction 6) or NioO (reaction 3). NioO could also further react with NioH to form the reduced Ni and water (reaction 7). No oxygen formation in the tail gas indicated that even a small amount of oxygen was produced by the decompositon of hydrogen peroxide, it could be dissociatively adsorbed on the Ni clusters by reaction 4 and then would be converted to water by reaction 7. This could also explain why the H2O2destruction activity of Ni/Al2O3catalyst was drastically increased in the presence of H2during the reaction.

Table 4 Composition of tail gas after hydrogenation of solution with different concentrations of hydrogen peroxide
4 Conclusions
The hydrogenation of methanol-containing effluent that was present in the exit stream from the epoxidation reactor in the HPPO process was tested with an aluminasupported nickel catalyst. At optimal reaction conditions (4.0 MPa, 150 ℃, molar ratio of H2/solution=40, and WHSV=2h-1), the acetaldehyde, hydrogen peroxide and nitro compounds in the solution could be completely eliminated. The hydrogenation process could remove acetone in the solution with high removal efficiency, but typically could achieve only a moderate removal rate of acetal and ketal. No significant changes were observed for the rest of the compounds that were present in the solution such as methanol, propylene glycol and propylene glycol monomethyl ether.
Compared with the decomposition process, hydrogen peroxide at different concentrations in the solution could be completely hydrogenated using the Ni catalyst and nooxygen was found in the reaction. The H2O2destruction activity of Ni/Al2O3catalyst was drastically increased in the presence of H2during the hydrogenation process because of the effective removal of chemisorbed O2by the H2over the Niospecies.
Increases in the reaction temperature, pressure, the quantity of hydrogen, and the catalyst amount will result in greater impurities removal rates, especially for the acetone and acetal and ketal. However, this outcome would also result in an increase in the production cost and a reduced process economics.
[1] Ma Qinggang. Production and consumption of propylene oxide[J]. Chemical Industry, 2011, 29 (2/3): 25-28 (in Chinese)
[2] Michael T D, Funada C. Marketing Research Report: Propylene Oxide[R]// Chemical Economics Handbook, IHS Chemical, 2012
[3] Richey W F. Chlorohydrins[M]//Kirk Orthmer. Encyclopedia of Chemical Technology, 4thEdition. New York: Wiley, 1994: 140
[4] Jorge E M. Chlorohydrin process: The United States, US 6043400[P]. 2000-05-28
[5] Kollar J. Epoxidation process. The United States, US 3351635[P]. 1967-11-07
[6] Buijnk J K F, van Vlaanderen J J M, Crocker M. Propylene epoxidation over titanium-on-silica catalyst—the heart of the SMPO process[J]. Catalysis Today, 2004, 93-95: 199-204
[7] Tsuji J; Yamamoto J, Ishino M, et al. Development of new propylene oxide process[J]. Sumitomo Kagaku, vol. 2006-I
[8] Cisneros M D, Holbrook M T, Ito L N. Hydrodechlorination process and catalyst for use therein: The United States, US 5476984[P]. 1995-12-19
[9] Parvulescu E. New mechanistic insight into the gold-based propene epoxidation[D]. Utrecht University, 2009
[10] Gelbein A P. Propylene oxide via direct oxidation of propylene[R]. PEP Review 98-3, 2000-09
[11] Clerici M G, Ingallina P. Epoxidation of lower olefins with hydrogen peroxide and titanium silicalite[J]. Journal of Catalysis, 1993, 140(1): 71-83
[12] Tullo A. Dow BASF to build propylene oxide: companies mark success in process with plans for plant in Belgium[J]. Chem Eng News, 2004, 82(36): 15
[13] Yang Hui, Jin Peng, Zheng Xiaoguang, et al. Preparation, characterization and propylene epoxidation performance of titanium silicalite[J]. Petroleum Processing and Petrochemicals, 2014, 45(5): 44-49 (in Chinese)
[14] Lin Min, Li Hua, Wang Wei, et al. The preparation of propylene oxide by propylene epoxidation with hydrogen peroxide in 1.0 kt/a pilot plant[J]. Petroleum Processing and Petrochemicals, 2013, 44(3): 1-5
[15] Yang Hui, Jin Peng, Zheng Xiaoguang, et al. Preparation of titanium silicalite, characterization and its application in propylene epoxidation with hydrogen peroxide[J]. Petroleum Processing and Petrochemicals, 2013, 44(6): 59-63
[16] Clerici M G, Bellussi G, Romano U. Synthesis of propylene oxide from propylene and hydrogen peroxide catalyzed by titanium silicalite[J]. Journal of Catalysis, 1991, 129(1): 159-167
[17] Capel-Sanchez M C, Campos-Martin J M, Fierro J L G. Influence of solvent in the synthesis steps of titanium-supported amorphous silica epoxidation catalysts[J]. Journal of Catalysis, 2003, 217(1): 195-202
[18] Capel-Sanchez M C, Campos-Martin J M, Fierro J L G. Impregnation treatments of TS-1 catalysts and their relevance in alkene epoxidation with hydrogen peroxide[J]. Applied Catalysis A: General, 2003, 246(1): 69-77
[19] Capel-Sanchez M C, Campos-Martin J M, Fierro J L G. Influence of the textural properties of supports on the behaviour of titanium-supported amorphous silica epoxidation catalysts[J]. Journal of Catalysis, 2005, 234(2): 488-495
[20] Klemm E, Dietzsch E, Schwarz T, et al. Direct gas-phase epoxidation of propene with hydrogen peroxide on TS-1 zeolite in a microstructured reactor[J]. Ind Eng Chem Res, 2008, 47(6): 2086-2090
[21] Brievaa G B, de Frutos-Escrigb M P, Martínb H, et al. Selective decomposition of hydrogen peroxide in the epoxidation effluent of the HPPO process[J]. Catalysis Today, 2012, 187(1): 168-172
[22] Landon P, Collier P J, Carley A F, et al. Direct synthesis of hydrogen peroxide from H2and O2using Pd and Au catalysts[J]. Physical Chemistry Chemical Physics, 2003(5): 1917-1923
[23] Brieva G B, de Frutos-Escrigb M P, Martínb H, et al. Selective hydrogenation of hydrogen peroxide in the epoxidation effluent of the HPPO process[J]. Catalysis Com-munications, 2012, 26: 83-87
[24] Haas T, Thiele G, Moroff G, et al. Process for the epoxidation of olefins: The United States, US 7141683[P]. 2006-11-28
[25] Gaikwad A G, Sansare S D, Choudhary V R. Direct oxidation of hydrogen to hydrogen peroxide over Pd-containing fluorinated or sulfated Al2O3, ZrO2, CeO2, ThO2, Y2O3and Ga2O3catalysts in stirred slurry reactor at ambient conditions[J]. Journal of Molecular Catalysis A: Chemical, 2002, 181(1/2): 143-149
[26] Samanta C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process[J]. Applied Catalysis A: General, 2008, 350(2): 133-149
[27] Hasan M A, Zaki M I, Pasupulety L, et al. Promotion of the hydrogen peroxide decomposition activity of manganese oxide catalysts[J]. Applied Catalysis A: General, 1999, 181(1): 171-179
[28] Teshima N, Genfa Z, Dasgupta P K. Catalytic decomposition of hydrogen peroxide by a flow-through self-regulating platinum black heater[J]. Anal Chim Acta, 2004, 510(1): 9-13
[29] Li Y M. Principle of Industrial Catalysis[D]. Tianjin: Tianjin University Press, 1992 (in Chinese)
[30] Choudhary V R, Samanta C, Jana P. Decomposition and/or hydrogenation of hydrogen peroxide over Pd/Al2O3catalyst in aqueous medium: Factors affecting the rate of H2O2destruction in presence of hydrogen[J]. Applied Catalysis A: General, 2007, 332(1): 70-78
[31] Choudhary V R, Samanta C, Jana P. Formation from direct oxidation of H2and destruction by decomposition/hydrogenation of H2O2over Pd/C catalyst in aqueous medium containing different acids and halide anions[J]. Applied Catalysis A: General, 2007, 317 (2): 234-243
Core-Shell Zeolite Catalyst for Methylation of Toluene Developed by Hunan University
The Hunan University has devised a core-shell structured zeolite (HSM-5@silicalite-1), which can be used as the methylation agent for manufacturing PX through reaction of bromomethane with toluene (CN103708496A,2014-04-09). Taking into account the demerits related with the structure of currently used catalysts, the research team has designed the composite catalyst having active zeolite in the core side and inert zeolite in the shell side in order to regulate and modify the acidity on the zeolite catalyst surface and increase its para-xylene selectivity.
This catalyst is composed of HZSM-5 in its core and silicalite-1 in its shell to constitute a homogeneous and compact shell packed with high-purity highly dispersed regular particles characteristic of excellent catalytic activity and para-xylene selectivity and stability. In a practical example, the catalyst core consisting of HZSM-5 with a Si/Al ratio of 75 after being dispersed in the tetrapropylammonium hydroxide solution is subject to ageing with ethyl silicate, crystallization and calcination to form the HSM-5@silicalite-1 with a shell thickness of 40 nm. This zeolite catalyst can be used in the reaction of toluene with bromomethane serving as the methylation agent for manufacturing xylene. At a temperature of 400℃ and a toluene weight hourly space velocity of 2 h-1, the toluene conversion can reach 52% in one hour with a para-xylene selectivity of 72%.
date: 2015-03-09; Accepted date: 2015-05-09.
Cheng Ke, Telephone: +86-379-66996375; E-mail: chengk.lysh@sinopec.com.

- 中国炼油与石油化工的其它文章
- Optimization of High-Gravity Chelated Iron Process for Removing H2S Based on Response Surface Methodology
- Studies on the Hydrogenation of Acetonitrile over Fresh Mo2C/γ-Al2O3Catalyst by In-situ IR Spectroscopy
- Synthesis of Biodiesel Using ZrO2Polycrystalline Ceramic Foam Catalyst in a Tubular Reactor
- Investigation of Swelling and Dissolution Process of Natural Rubber in Aromatic Oil
- Secondary Crystallization of Na2CO3-Modified HZSM-5 Zeolites with Tetrapropylammonium Hydroxide and Their Catalytic Performance in Thiophene Alkylation Reaction
- Lubricant Biodegradation Enhancers: Designed Chemistry and Engineered Technology