
自噬相关基因Atg4B及LC3-Ⅱ在肌萎缩侧索硬化症转基因鼠海马中的表达及意义
2015-06-12 12:36周风华刘焕彩陈燕春蒲雷东王巧真接琳琳丁昊宇
中国老年学杂志 2015年12期
周风华 刘焕彩 陈燕春 鞠 杰 蒲雷东 王巧真 接琳琳 丁昊宇
(潍坊医学院病理学教研室,山东 潍坊 261053)
自噬相关基因Atg4B及LC3-Ⅱ在肌萎缩侧索硬化症转基因鼠海马中的表达及意义
周风华 刘焕彩1陈燕春2鞠 杰2蒲雷东2王巧真2接琳琳2丁昊宇2
(潍坊医学院病理学教研室,山东 潍坊 261053)
目的 观察自噬相关基因Atg4B及LC3-Ⅱ在肌萎缩侧索硬化症(ALS)转基因鼠海马组织中的表达变化,探讨细胞自噬机制在ALS发病中的作用及意义。方法 选择SOD1-G93A转基因鼠进行试验,分别于发病早、中、晚期,通过RT-PCR、免疫荧光检测小鼠海马组织中Atg4B、LC3-Ⅱ在mRNA、蛋白水平的表达及变化。结果 与野生型鼠相比较,转基因鼠的Atg4B在mRNA及蛋白水平表达均下调;LC3-Ⅱ在蛋白水平表达上调,在mRNA水平变化不明显。结论 自噬相关基因Atg4B及LC3-Ⅱ参与了ALS发病,ALS海马组织的病变与自噬异常有关。
自噬;ALS;Atg4B;LC3-Ⅱ
肌萎缩侧索硬化症(ALS)是一种选择性上、下运动神经元进行性变性为主要特征的神经退行性疾病,临床主要表现为进行性全身肌肉无力,多数患者在发病3~5年内死于呼吸衰竭〔1〕。除运动系统功能障碍外,部分ALS患者可出现明显的认知功能等行为学损害〔2〕因而ALS的发展可能与海马结构有关〔3〕。细胞自噬是真核细胞特有的生命现象〔4〕,参与人体的多种生理及病理过程。Atg4B及LC3-Ⅱ是两个重要的自噬相关基因,参与了自噬体的形成。Morimoto等〔5〕研究发现,ALS的发病与细胞自噬有关,而自噬基因Atg4B及LC3-Ⅱ在ALS海马中的表达变化及作用尚无明确报道。本实验通过检测Atg4B及LC3-Ⅱ在ALS转基因鼠海马组织中的表达变化,探讨细胞自噬在ALS发病中的作用。
1 材料与方法
1.1 动物模型及标本处理 野生型及突变型SOD1-G93A转基因小鼠购自美国Jackson Laboratories。该动物模型携带有突变的SOD1基因,能够模拟人类ALS的疾病进展及临床表现,是研究ALS疾病的理想动物模型。严格按转基因鼠饲养条件进行饲养,成年鼠按雌雄比1∶1进行合笼,出生后的小鼠约30 d后,提取鼠尾DNA,基因扩增进行野生型及突变型小鼠鉴定。选取突变型成年鼠18只,随机分为发病早期(生后约95 d)、发病中期(出生后约108 d)、发病晚期(出生后约122 d)。每个时间点选3只鼠进行4%多聚甲醛灌注固定,剥离大脑,30%蔗糖脱水,冰冻切片,进行免疫荧光检测;每个时间点选3只鼠拉颈处死,剥离海马,提取总RNA并反转录合成cDNA,进行RT-PCR检测;以相同数量的同窝野生型鼠作为对照组。
1.2 主要试剂 兔抗多克隆抗体Atg4B,anbo公司;LC3-Ⅱ,Abcam公司;Cy3标记的羊抗兔IgG,美国Jackson Immuno Research Laboratoris;RT-PCR试剂盒,北京天根生化科技有限公司;OligdT,美国Promega公司;5×逆转录Buffer,美国Promega公司;M-MlV逆转录酶,美国Promega公司。
1.3 免疫荧光标记 将冰冻切片室温晾片2 h,滴加10%山羊血清,37℃封闭40 min,分别滴加兔抗多克隆抗体Atg4B、LC3-Ⅱ(浓度均为1∶100),4℃孵育过夜,滴加Cy3标记的羊抗兔IgG荧光二抗(浓度为1∶400),避光37℃孵育30 min,滴加终浓度为10 μg/ml的Hoechst33258,避光37℃孵育15 min,防淬灭荧光封片剂封片,用荧光显微镜观察Atg4B及LC3-Ⅱ表达情况。实验过程中的冲洗缓冲液均为0.01 mol/L的PBS缓冲液。
1.4 RT-PCR (1)总RNA的提取:取大脑海马组织,加入500 μl TRIzol,冰上充分匀浆,具体步骤参照文献〔6〕。(2)逆转录反应。(3)PCR扩增反应:Atg4B上游引物:5′TACAGCATTTTCACAGAGAAGGACG 3′,Atg4B下游引物:5′CTCCAGCAGGGAACCCATTAC3′。LC3-Ⅱ上游引物:5′CCCAGTGATTATAGAGCGATACAAG3′,LC3-Ⅱ下游引物:5′TGTCTCCTGCGAGGCATAAAC3′。以1 μl逆转录产物为模板,2×Taq PCR Mastermix 10 μl。Atg4B、LC3-Ⅱ上下游引物分别各0.5 μl,β-actin上游引物、下游引物各0.3 μl,分别组成PCR反应体系。PCR仪中运行以下程序:94℃预变性3 min,94℃变性30 s,57℃退火30 s,72℃延伸30 s,循环30次,72℃延伸5 min后冷却至4℃。
1.5 统计学处理 应用IPP5.1图像分析软件进行统计学分析。测量Atg4B及LC3-Ⅱ免疫荧光图片阳性细胞累积光密度(IOD)值。测量RT-PCR显示的mRNA扩增带与β-actin扩增带的IOD值。用SPSS20.0软件行独立样本t检验。
2 结 果
2.1 Atg4B在海马组织中的表达 免疫荧光检测显示,在野生型及突变型小鼠海马组织中,均检测到Atg4B阳性细胞,阳性细胞主要分布在海马的DG区,CA可见少量阳性细胞。与野生型小鼠比较,在发病的早期,突变型小鼠Atg4B阳性细胞累积光密度变化不明显,而在发病中、晚期,突变型小鼠Atg4B阳性细胞累积光密度在DG区明显降低(68.5±5.2 vs 32.3±2.6,P<0.05),CA区阳性细胞累积光密度变化不明显(P>0.05)。RT-PCR结果显示,与野生型小鼠比较,在发病的早(61.2±5.7 vs 58.2±7.2)、中(60.1±5.2 vs 38.5±4.7)、晚期(59.5±4.2 vs 39.7±3.5),突变型小鼠Atg4B mRNA水平明显下调(P<0.05),见图1,图2。
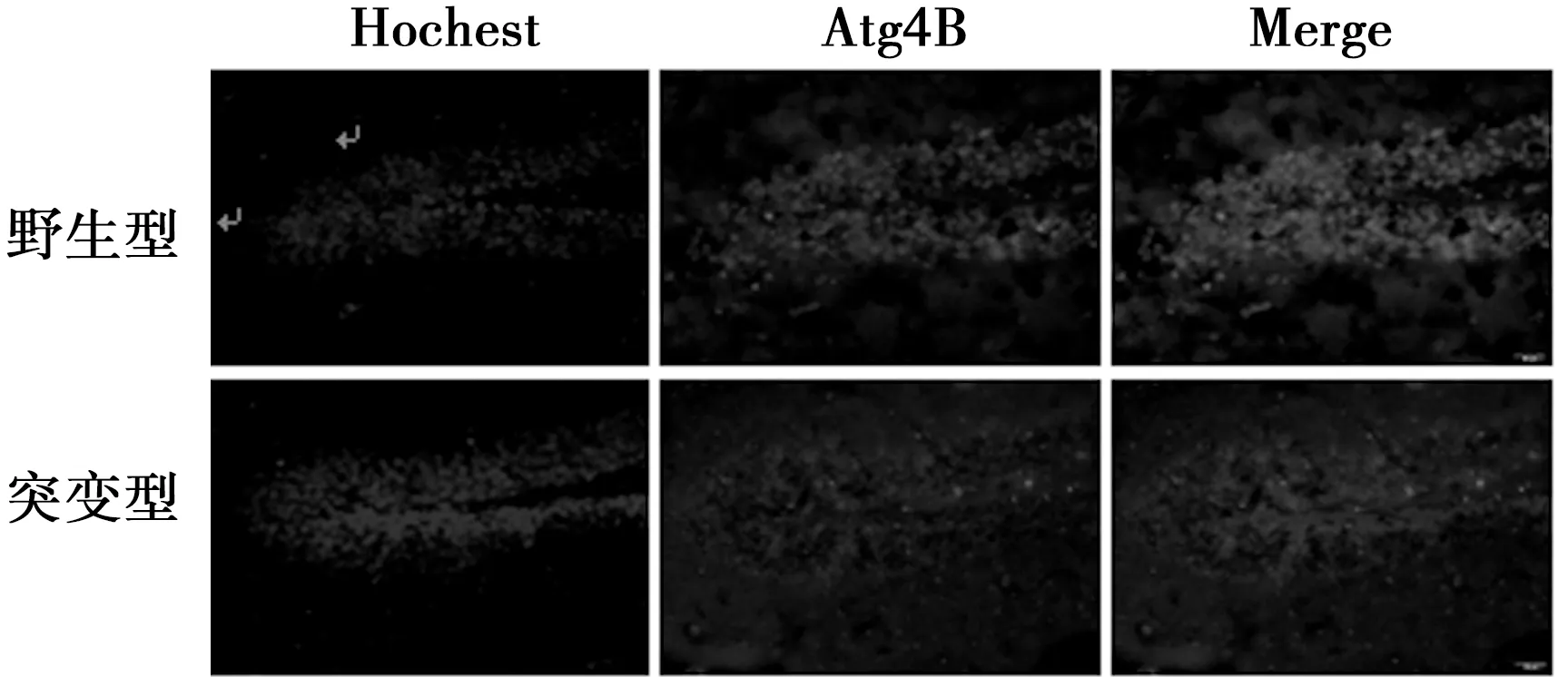
图1 Atg4B在突变型小鼠122 d海马组织中的表达(标尺=50 μm)
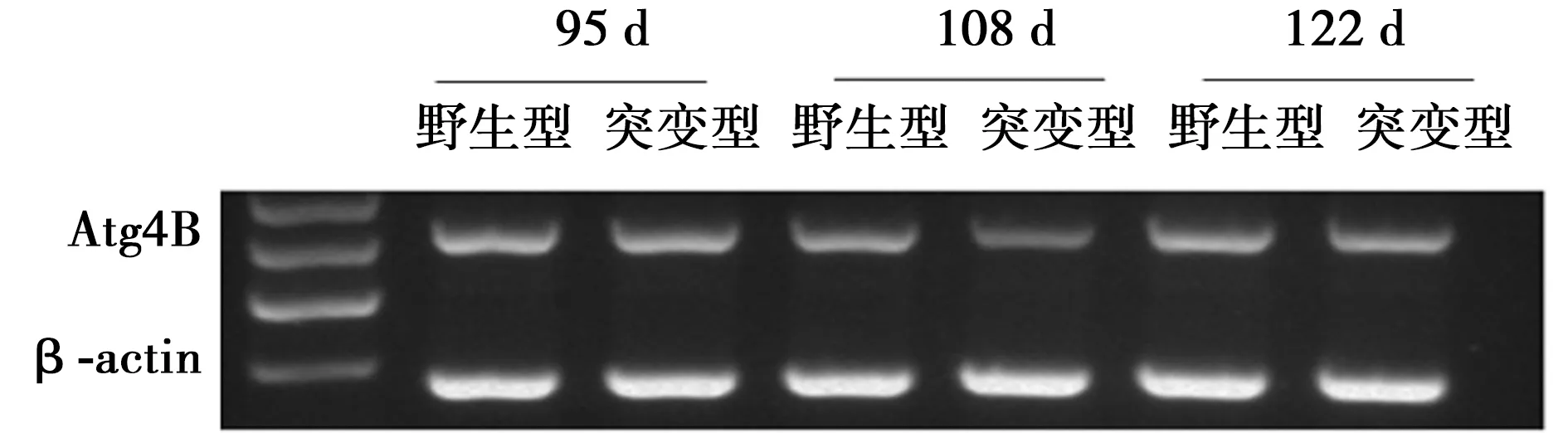
图2 Atg4B在突变型小鼠发病早、中、晚期mRNA表达变化
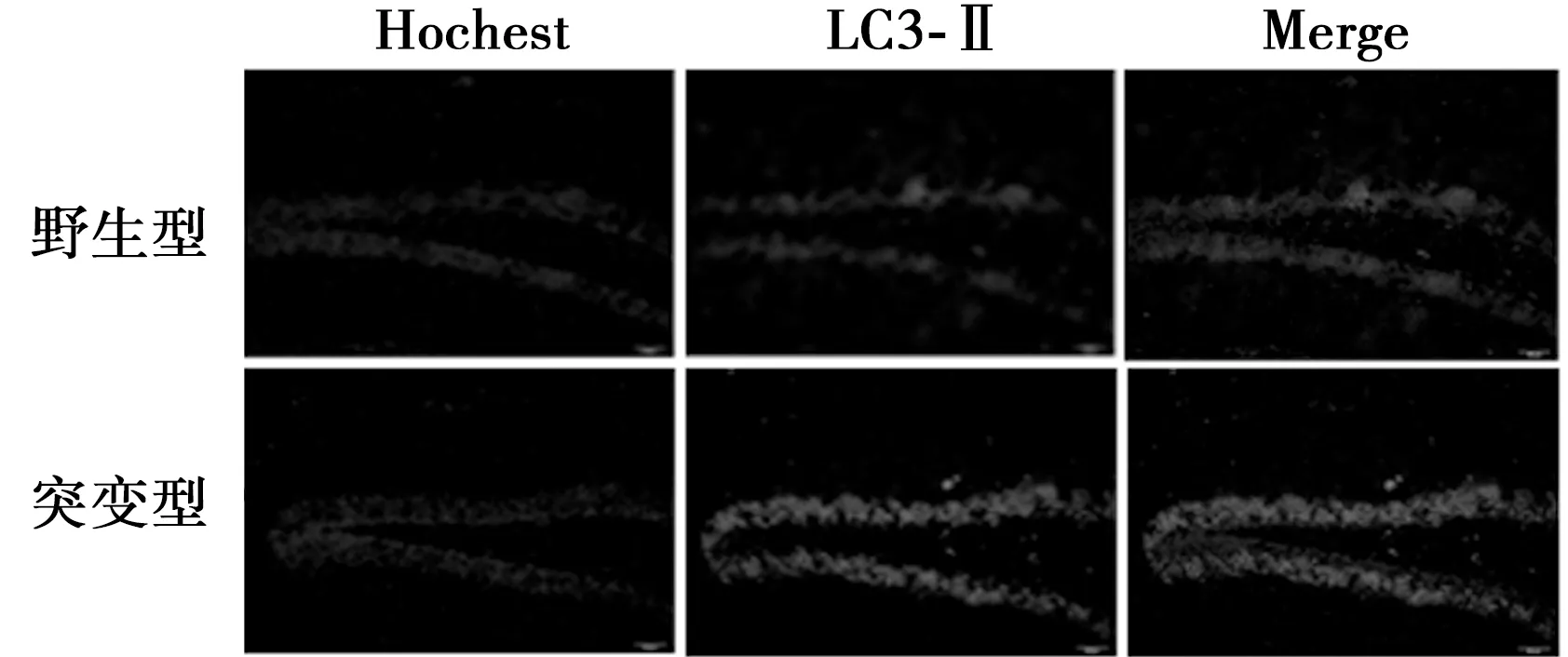
图3 LC3-Ⅱ在突变型小鼠122 d海马组织中的表达(标尺=50 μm)
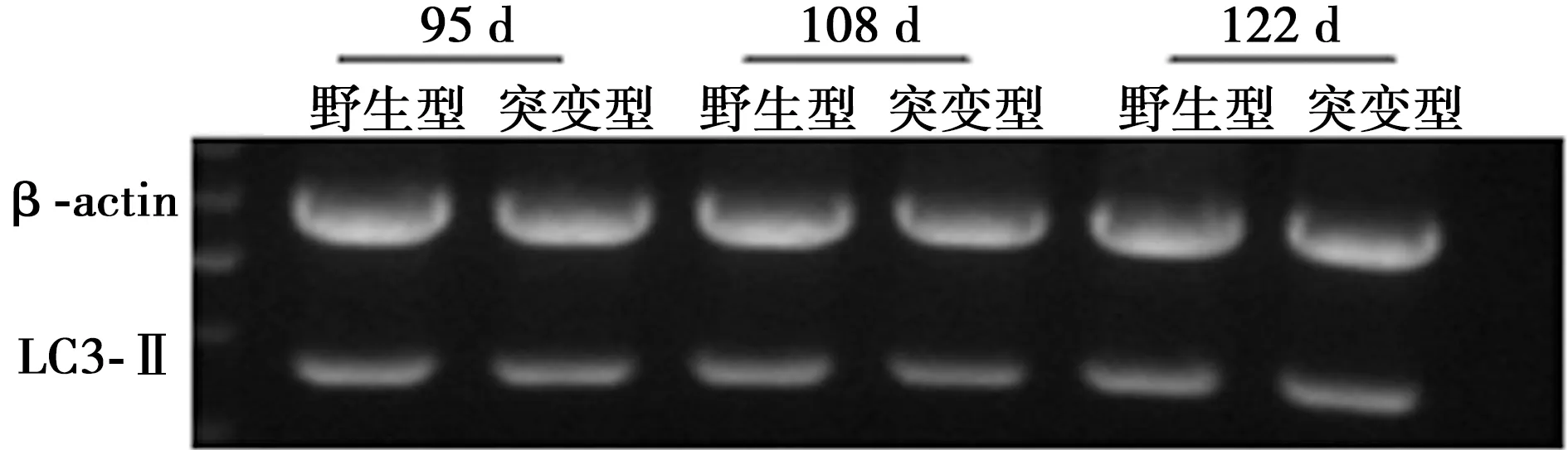
图4 LC3-Ⅱ在突变型小鼠发病早、中、晚期mRNA表达变化
2.2 LC3-Ⅱ在海马组织中的表达呈上调趋势 免疫荧光检测显示,在野生型及突变型小鼠海马组织中,均检测到LC3-Ⅱ阳性细胞,阳性细胞主要分布在海马的DG区。与野生型小鼠比较,在发病的早期,突变型鼠LC3-Ⅱ阳性细胞累积光密度变化不明显,在发病的中、晚期,突变型鼠LC3-Ⅱ阳性细胞累积光密度明显增加(62.5±6.1 vs 35.3±4.5,P<0.05)。RT-PCR结果显示,与野生型小鼠比较,在发病的早(36.5±4.2 vvs 34.1±5.3)、中(38.3±5.1 vs 39.1±4.3)、晚期(40.5±5.6 vs 39.8±4.2),突变型小鼠LC3-Ⅱ mRNA水平变化不明显(P>0.05),见图3,图4。
3 讨 论
自噬是真核细胞特有的生命现象〔4〕,参与人体的多种生理及病理过程。有研究报道,自噬与阿尔茨海默病(AD)、亨廷顿舞蹈病(HD)和帕金森病(PD)等多种神经退行性疾病有关〔6〕。在这些神经退行性疾病中,自噬可能通过清除异常折叠的蛋白而延缓疾病的进展。尸体剖验及动物实验均证实在ALS脊髓组织中自噬小体的数量增加〔5〕,提示在ALS发病中自噬被激活,但目前自噬的激活机制及意义尚未完全阐明。
微管相关蛋白轻链3(LC3)是哺乳动物细胞中酵母8(Atg8)基因的同源物,参与细胞的自噬过程。LC3有Ⅰ型和Ⅱ型之分,当自噬尚未发生时,LC3以可溶性的LC3-Ⅰ存在,当自噬发生时,LC3与自噬泡膜表面的磷脂酰乙醇胺(PE)结合,转化成脂溶性的LC3-Ⅱ〔7〕。LC3-Ⅱ的含量多少与自噬泡的数量成正比,因此LC3-Ⅱ的表达强度与自噬活性密切相关,是自噬检测的标记〔8〕。LC3-Ⅱ与多种神经退行性疾病有关,在AD转基因鼠及β-淀粉样蛋白作用的原代皮质神经元中LC3-Ⅱ表达上调。而亨廷顿蛋白的突变导致了骨骼肌中LC3-Ⅱ的合成增加〔9〕。Morimoto等〔5〕研究发现,在ALS发病期,LC3-Ⅱ在SOD1G93A转基因小鼠模型脊髓中是上调的,表明LC3-Ⅱ诱导的自噬激活可能参与了ALS发病。我们的实验结果显示,在ALS发病期,海马组织中LC3-Ⅱ在mRNA及蛋白水平均明显上调,由此提示,ALS海马组织的病变与自噬异常有关。最近,一个大规模的蛋白质组学实验揭示在人自噬过程中,LC3-Ⅱ和它的直系同源物组成了约67种蛋白的网络〔10〕。而LC3-Ⅱ调控自噬途径的机制可能与蛋白质之间的相互作用有关。Atg4是一种半胱氨酸蛋白激酶,能够可逆性的催化LC3形成脂质化及去脂质化LC3。在哺乳动物中,有四种Atg4的同系物,包括Atg4A、Atg4B、Atg4C及 autophagin-4,其中Atg4B对LC3-Ⅱ有明显的特异性及高效性。研究发现,非活化Atg4B的高表达可以通过抑制LC3-I的脂质化而明显抑制自噬体的形成〔11〕,由此提示,Atg4B和LC3通过之间的相互作用参与了细胞的自噬过程。目前,在ALS发病中Atg4B和LC3-Ⅱ是否参与了海马组织病变尚未见报道。我们的研究结果显示,Atg4B和LC3-Ⅱ均主要在海马组织的DG区表达,但是表达趋势相反,在ALS发病期,海马组织中Atg4B的表达下调,而LC3-Ⅱ的表达明显上调,提示Atg4B和LC3-Ⅱ在ALS发病期海马组织的病变中发挥了作用,而且两者之间还可能通过一定的调控机制共同参与其中。同时,我们还发现,在ALS转基因鼠的海马组织中,自噬相关基因LC3-Ⅱ在蛋白水平明显上调,但是在mRNA水平变化并不明显,因此,LC3-Ⅱ在蛋白水平的表达可能存在更复杂的分子机制。这些分子机制有待于我们进一步研究和探讨。
1 Renton AE,Chiò A,Traynor BJ.State of play in amyotrophic lateral sclerosis genetics〔J〕.Nat Neurosci,2014;17(1):17-23.
2 Phukan J,Elamin M,Bede P,etal.The syndrome of cognitive impairment in amyotrophic lateral sclerosis:a population-based study〔J〕.J Neurol Neurosurg Psychiatry,2012;83(1):102-8.
3 Takeda T,Uchihara T,Arai N,etal.Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment:distinction from Alzheimer disease〔J〕.Acta Neuropathol,2009;117(1):35-44.
4 Klionsky DJ,Emr SD.Autophagy as a regμlated pathway of cellμlar degradation〔J〕.Science,2000;290(5497):1717-21.
5 Morimoto N,Nagai M,Ohta Y,etal.Increased autophagy in transgenic mice with a G93A mutant SOD1 gene〔J〕.Brain Res,2007;1167:112-7.
6 Kuusisto E,Salminen A,Alafuzoff I.Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies〔J〕.Neuroreport,2001;12:2085-90.
7 Bjorkoy G,Lamark T,Brech A,etal.p62/SQSTM1forms protein aggregates degraded by autophagy and has a protective effect on Huntingtin-induced cell death〔J〕.J Cell Biol,2005;171:603-14.
8 Nakaso K,Yoshimoto Y,Nakano T,etal.Transcriptional activation of p62/A170/ZIP during the formation of the aggregates:possible mechanisms and the role in Lewy body formation in Parkinson′s disease〔J〕.Brain Res,2004;1012:42-51.
9 Sasaki S.Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis〔J〕.J Neuropathol Exp Neurol,2011;70(5):349-59.
10 Geng J,Klionsky DJ.The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy.Protein modifications:beyond the usual suspects′ review series〔J〕.EMBO Rep,2008;9(9):859-64.
11 Kabeya Y,Mizushima N,Ueno T,etal.LC3,a mammalian homologue of yeast Apg8p,is localized in autophagosome membranes after processing〔J〕.EMBO J,2000;19(21):5720-8.
〔2014-09-19修回〕
(编辑 郭 菁)
国家自然科学基金青年基金(81401066);山东省自然科学基金(ZR2012HQ021);山东省教育厅课题(J11LF16,J12LK51,J13LK05);山东省医药卫生科技发展计划项目(2013W-S0279);潍坊市科学技术发展计划项目(201302089,2013-01074)
陈燕春(1979-),女,副教授,博士,主要从事神经发育与神经退行性疾病相关研究。
周风华(1977-),女,副教授,硕士,主要从事神经病理研究。
R744.8
A
1005-9202(2015)12-3216-03;
10.3969/j.issn.1005-9202.2015.12.013
1 潍坊医学院临床学院 2 潍坊医学院组织胚胎学教研室
猜你喜欢
哈尔滨理工大学学报(2022年1期)2022-05-10
国际眼科杂志(2022年2期)2022-02-18
中国病理生理杂志(2020年3期)2020-04-03
野生动物学报(2020年1期)2020-02-21
中国病理生理杂志(2018年9期)2018-09-27
江苏农业科学(2017年15期)2018-02-06
中国高原医学与生物学杂志(2017年4期)2017-03-08
癌症进展(2015年5期)2015-07-26
中国现代医生(2015年5期)2015-03-31
火炸药学报(2015年1期)2015-03-05
