
转化生长因子β1通过Smad3调控补体应答基因32转录的分子机制
2015-06-09 06:49:10沈云琳钮小玲刘华杰匡新宇黄文彦
肾脏病与透析肾移植杂志 2015年5期
沈云琳 钮小玲 刘华杰 孙 蕾 匡新宇 黄文彦
转化生长因子β1通过Smad3调控补体应答基因32转录的分子机制
沈云琳 钮小玲 刘华杰 孙 蕾 匡新宇 黄文彦
目的:前期工作发现补体应答基因32(RGC-32)是转化生长因子β(TGF-β)诱导的肾小管上皮细胞-间充质转化(EMT)过程中Smad信号下游的关键调控分子。本研究拟进一步明确TGF-β诱导肾小管EMT过程中RGC-32的转录调控机制。 方法:体外培养NRK-52E细胞,利用荧光素酶报告基因实验检测RGC-32启动子活性以确定Smad结合元件(SBE)是否为TGF-β诱导的RGC-32转录调控位点;通过凝胶迁移率实验(EMSA)明确与之结合的转录调控分子。 结果:(1)转染野生型SBE片段的NRK-52E细胞荧光素酶活性明显增高,而转染突变的SBE片段的细胞无此表现,表明SBE是调控TGF-β诱导的RGC-32转录启动的关键位点。(2)合成32P标记的包含SBE的寡核苷酸探针行EMSA可见TGF-β诱导的核蛋白与探针复合物生成,且加入包含SBE序列的竞争片段可以抑制此核蛋白-探针复合物的形成,表明有蛋白与SBE结合并调控RGC-32的转录。(3)当加入不同的Smad抗体行超迁移实验发现Smad2和Smad3抗体可以和复合物相互作用,表明Smad2和Smad3可能是TGF-β诱导的转录复合物的组成部分。(4)将Smad2和Smad3分别与p-1500RGCluc共同转染NRK-52E细胞,Smad3可以显著增加RGC-32启动子活性,表明Smad3是TGF-β诱导的转录复合物的组成部分,而不是Smad2。 结论:在TGF-β诱导肾小管EMT过程中,Smad3通过与RGC-32启动子区SBE结合,从而调控肾小管EMT过程。
补体应答基因32 上皮-间充质转化 转化生长因子β 转录调控
慢性肾脏病(CKD)一直是困扰全世界的难题,并严重威胁着人类健康。而肾小管间质纤维化的程度与肾功能下降及CKD的进程密切相关[1-3]。肾小管间质纤维化的一个重要过程是上皮细胞-间充质转化(EMT)[3-5],主要表现为上皮细胞的表型和功能丢失,上皮细胞标志物如E-钙黏蛋白表达降低,而间质细胞标志物如N-钙黏蛋白表达增强,并且获得产生细胞外基质(ECM)的能力,如胶原蛋白,纤维连接蛋白和α平滑肌肌动蛋白(α-SMA)表达增加[6-7]。现已明确转化生长因子β(TGF-β)信号转导通路在肾小管EMT过程中起关键作用,而Smad蛋白是TGF-β信号转导通路中将信号由胞质转导至细胞核的中介分子[8]。然而,TGF-β诱导EMT的详细分子调控机制尚有待进一步阐明。我们先前的研究发现,补体应答基因32(RGC-32)作为TGF-β下游关键调控分子,参与了Smad信号通路介导的肾小管上皮细胞EMT过程[9]。在TGF-β诱导的神经嵴细胞平滑肌细胞分化中,Smad2和PEA3相互作用共同调节RGC-32的转录活性[10]。最近,Guo等[11]发现RGC-32必须与Smad3相互作用诱导人肾小管细胞EMT。但Smad3调控RGC-32的分子机制尚有待明确,为此,本研究通过体外培养大鼠肾小管上皮细胞(NRK-52E)明确Smad3调控RGC-32转录的分子机制。
材料和方法
细胞培养和试剂 大鼠肾小管上皮细胞(NRK-52E)细胞株购自ATCC(American Type Culture Collection)。细胞培养用含5%胎牛血清(GIBCO公司)Dulbecco’s modified Eagle’s medium(DMEM)培养基(GIBCO公司),37 ℃,95%空气和5%二氧化碳细胞培养箱培养。将NRK-52E细胞按3×105/孔接种至六孔板中培养至80%融合,换无血清DMEM培养基培养6h,然后按实验需要予TGF-β1(5 ng/ml)刺激不同时间后收集细胞用于检测。Smad1、Smad2、Smad3、Smad4和Smad5抗体(兔多抗IgG)购自Santa Cruz公司。Smad1、Smad2、Smad3、Smad4和Smad5表达质粒[12]及小干扰RNA(small interfering RNA,siRNA)均购自于OriGene公司。TGF-β1来源于R&D Systems公司。
RGC-32启动子质粒构建 参照文献10,即利用大鼠基因组DNA为模板,通过PCR方法扩增RGC-32启动子DNA,并克隆至经KpnI/Bgl II限制性内切酶消化的含荧光素酶报告载体pGL3-basic(Promega)位点之间。pGL3-RGC-32(-1 500)所包含的调控区域位于RGC-32基因-1 500 bp至22 bp处。引物设计为:正向:5>A ̄A ̄A ̄A ̄G ̄G ̄T ̄A ̄C ̄C ̄G ̄C ̄A ̄T ̄G ̄C ̄C ̄A ̄C ̄A ̄T ̄T ̄C ̄T ̄G ̄A ̄C ̄A ̄C ̄C<3;反向:5>A ̄A ̄A ̄A ̄A ̄G ̄A ̄T ̄C ̄T ̄C ̄G ̄C ̄T ̄A ̄G ̄C ̄T ̄A ̄C ̄T ̄C ̄G ̄C ̄G ̄T ̄G ̄A ̄G<3。pGL3-RGC-32(-1 500)突变质粒采用Quikchange Multi Site-Directed Mutagenesis Kit突变试剂盒(Stratagene 公司)构建,按试剂盒说明书使用。Smad结合位点(SBE)序列为GTCTGGAC(-1 344 bp至-1 337 bp)突变为GTtatGAC。
转染和荧光素酶报告基因实验 将NRK-52E细胞以2×105/孔接种于12孔板中培养至细胞80%融合,然后严格按照说明书用LipofectAMINE 2000转染细胞,所有样品均设三个复孔。50%的转染细胞观察到有增强的绿色荧光蛋白质粒转染。用荧光酶标仪(BioTek instruments)检测荧光素酶活性。实验均重复5次,结果取均值。
凝胶迁移率(EMSA)实验 首先提取NRK-52E细胞核蛋白,并测定浓度,然后标记寡核苷酸探针。EMSA实验方法[10,13]如下:(1)结合反应:将DNA结合反应液15 μl和核蛋白提取物5 μg加入EP管内冰浴15 min,加入已标记的寡核苷酸双链1 μl,25℃孵育30 min,加入4 μl加样缓冲液,混匀后加样。(2)特异性竞争抑制实验:将DNA结合反应液15 μl和核蛋白提取物5 μg加入EP管内冰浴15 min,加入50倍未标记的同样的寡核苷酸双链,25℃孵育20 min,加入已标记的寡核苷酸双链1 μl,25℃孵育30 min,加入4 μl加样缓冲液,混匀后加样。(3)超迁移实验:在结合反应的基础上再加入了1 μg Smad抗体,25℃孵育30 min,加入4 μl加样缓冲液,混匀后加样。蛋白-DNA 复合物用5%非变性聚丙烯酰胺凝胶分离并行放射自显影。突变探针的突变位点与之前突变荧光素酶质粒的描述相同。
统计学分析 结果采用SPSS 10.0统计软件包进行统计,所有数值均用均值±标准差表示。数据分析两组间比较采用ANOVA,P<0.05为差异有统计学意义。
结 果
TGF-β诱导RGC-32转录调控位点的验证 SBE序列为GTCTGGAC,位于RGC-32基因转录起始位点上游-1 344 bp至-1 337 bp。Huang等[10]报道,在神经嵴细胞向平滑肌细胞分化过程中,Smad2和PEA3共同作用调控RGC-32的转录,而SBE为RGC-32转录调控的结合位点。为了验证SBE是否在TGF-β诱导的肾小管上皮NRK-52E细胞RGC-32转录中同样起调控作用,我们设计了pGL3-RGC-32(-1 500)序列的部分核苷酸突变位点(图1A),然后将野生型和突变序列分别转染至NRK-52E细胞,TGF-β1刺激后行荧光素酶活性测定。结果发现突变的SBE较野生型的荧光素酶活性明显降低(图1B),表明SBE是TGF-β诱导的RGC-32转录的调控位点。
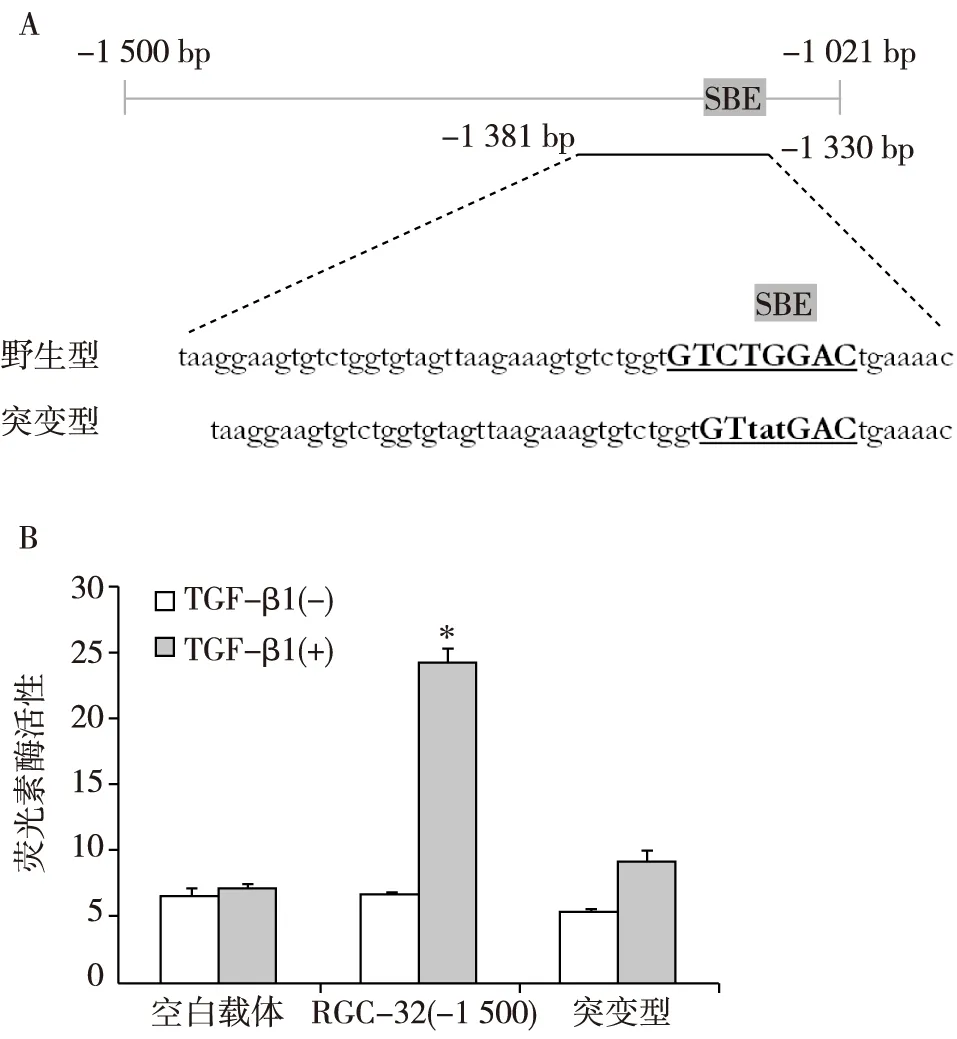
图1 TGF-β诱导RGC-32转录调控位点的验证SBE:Smad结合元件;TGF-β:转化生长因子β;RGC-32:补体应答基因32;A:RGC-32基因转录起始位点上游-1 500 bp至-1 021 bp中包含SBE序列,野生型中下划线部分为野生型SBE序列,而突变型中下划线部分为突变的SBE序列,图中数字为RGC-32基因转录起始位点上游相应碱基数;B:将野生型和突变型转染NRK-52E细胞20h,然后细胞用TGF-β1(5 ng/ml)刺激18h后测定荧光素酶活性;*:与空白载体组比较,P<0.05
TGF-β诱导的核蛋白与SBE结合形成复合物 为了观察TGF-β诱导的核蛋白与SBE相互作用,TGF-β1刺激NRK-52E细胞后,将核蛋白提取物与32P标记的包含SBE的DNA寡核苷酸探针孵育,然后行EMSA。结果发现,TGF-β1刺激后30 min、2 h和6 h均存在TGF-β诱导的核蛋白与探针复合物生成(图2A)。当加入包含SBE的野生型DNA序列的竞争片段可抑制此核蛋白-探针复合物的形成,而加入包含突变的SBE序列的竞争片段则没有此作用(图2B)。

图2 TGF-β诱导的核蛋白与SBE结合形成复合物TGF-β:转化生长因子β;SBE:Smad结合元件;EMSA:凝胶迁移率试验;A:NRK-52E细胞用TGF-β(5 ng/ml)刺激后,其核蛋白提取物与32P标记的SBE探针孵育,行EMSA结果;B:在核蛋白提取物与探针孵育时,再加入包含野生型SBE的DNA片段或包含突变SBE的DNA片段作为竞争物,行竞争性EMSA实验
TGF-β诱导的与SBE结合的核蛋白 上述实验证明有TGF-β诱导的核蛋白与探针复合物生成,但仍不清楚蛋白质性质。于是我们在上述实验的基础上又分别加入不同的Smad抗体行超迁移实验,以了解Smad蛋白是否是TGF-β诱导的核蛋白-探针复合物的组成成分。通过超迁移实验发现,加入Smad2或Smad3抗体的转录复合物出现迁移延迟,但Smad1、Smad4 和Smad5抗体则没有,表明Smad2和Smad3抗体可以和复合物相互作用,可能是TGF-β诱导的转录复合物的组成成分。

图3 TGF-β诱导的与SBE结合的核蛋白TGF-β:转化生长因子β;SBE:Smad结合元件;EMSA:凝胶迁移率试验;在上述EMSA实验基础上,将核蛋白提取物与1.0 μg的Smad1(S1)、Smad2(S2)、Smad3(S3)、Smad4(S4)和Smad5(S5)放在冰上孵育0.5 h,用正常羊IgG作为对照(C),然后行超迁移EMSA,观察转录复合物有无迁移延迟
Smad3是TGF-β诱导RGC-32转录活性的关键分子 以上实验可以明确Smad2和(或)Smad3参与了TGF-β诱导RGC-32启动的复合物的组成,但是何种Smad是RGC-32转录启动的关键我们还不清楚。我们将pGL3-RGC-32(-1 500)质粒和Smad1、Smad2、Smad3、Smad4和Smad5表达质粒共同转染到NRK-52E细胞,然后用TGF-β刺激。荧光素酶报告基因测定显示Smad1和Smad5对RGC-32转录活性没有影响,但Smad2、Smad3和Smad4明显上调RGC-32转录活性(图4A);而当转染不同的Smad siRNA后,转染Smad3 siRNA的细胞RGC-32转录活性下降(图4B)。将Smad2和Smad3表达质粒分别或一起与RGC-32(-1 500)质粒共同转染NRK-52E细胞后发现,转染Smad3后RGC-32的转录活性明显上调(图4C)。

图4 Smad3是TGF-β诱导RGC-32转录活性的关键分子TGF-β:转化生长因子β;RCG-32:补体应答基因32;A:将RGC-32(-1 500)质粒分别与Smad1、Smad2、Smad3、Smad4和Smad5表达质粒共同转染NRK-52E细胞20h,然后用TGF-β1(5 ng/ml)或TGF-β1(-)刺激18h,测定荧光素酶活性;B:将RGC-32(-1 500)质粒分别与Smads siRNA (Smad-S)或对照(siCtrl)共同转染NRK-52E细胞48h,然后用TGF-β1(5 ng/ml)刺激18h,测定荧光素酶活性;C:将Smad2和Smad3表达质粒分别或一起与RGC-32(-1 500)共同转染NRK-52E细胞20h,然后用TGF-β1(5 ng/ml)或TGF-β1(-)刺激18h,测定荧光素酶活性;*:与TGF-β1(-)组比较,P<0.05
讨 论
TGF-β是转化生长因子超家族中的主要成员,可以通过受体表面的受体信号转导途径调节细胞的增殖、分化和凋亡,对细胞外基质的合成、创伤的修复、免疫功能等有重要的调节作用。在CKD中TGF-β是肾小子,TGF-β信号的转导分为Smad通路和非Smad通路。许多研究已经揭示,TGF-β信号通路和Smad蛋白包括Smad2、Smad3、Smad4和Smad7,在肾脏纤[12,14-15]。TGF-β配体和丝氨酸/苏氨酸蛋白激酶Ⅱ型受体结合并使其活化,活化的Ⅱ型受体募集并结合Ⅰ型受体,形成异源三聚体,使Ⅰ型受体磷酸化,活化的Ⅰ型受体特异性地与Smad蛋白相互作用,并将信号转入细胞核[16]。
Smad蛋白可分为三类,分别为受体激活型Smad(Smad2和Smad3)、公共介体型Smad(Smad4)和抑制型Smad(Smad7)。活化的Ⅰ型受体特异性地与受体激活型Smad2和Smad3相互作用使其活化,活化的Smad2/3还必须与公共介体型Smad4结合,Smad4协助活化的Smad2/3向核内转移,并保持其转录活性,转入核内的Smad2/3通过与DNA结合或与其他转录因子共同作用而参与转录调节[8,16-17]。目前认为Smad2/3是肾脏纤维化时TGF-β1诱导的EMT过程中必需的[18],但是具体的机制仍不清楚。
RGC-32是一种重要的补体应答基因,首次由Badea等[19]克隆,被发现存在于多种人类组织中,包括心、脑、肝、骨骼肌、胎盘、肾脏和胰腺。在人类某些肿瘤组织中,如结肠癌组织中,RGC-32表达明显增高[20]。RGC-32还是周期素依赖激酶p34cdc2的调控分子,对细胞周期的激活起到重要作用[19,21]。我们前期研究发现RGC-32作为TGF-β下游Smad信号通路关键调控分子参与了TGF-β诱导的肾小管细胞EMT[9]。Verrecchia等[22]报道某些纤维发生基因(胶原蛋白)和EMT标志物(α-SMA和E-cadherin)依赖于Smad3。最近,Guo等[11]研究发现,RGC-32必须与Smad3相互作用并调控成纤维细胞标志物α-SMA、ECM和E-cadherin的表达。然而,Smad3如何调控RGC-32的分子机制仍不清楚。
本研究中,我们首次阐明在TGF-β诱导NRK-52E细胞EMT过程中,Smad3调控RGC-32基因转录活性。首先,通过荧光素酶报告基因实验和EMSA实验证实RGC-32基因启动子区上游的SBE序列(-1 344至-1 337 bp)为TGF-β诱导的核蛋白与RGC-32启动子相互作用的结合位点。第二,通过特异性抗体的识别发现,Smad2和Smad3抗体可以与TGF-β诱导的与RGC-32启动子结合的转录因子复合物结合,因此Smad2/3可能是复合物的组成成分。由于Smad4是公共介体型Smad,其可协助活化的Smad2/3向核内转移,其本身不是复合物的组成成分,故无超迁移条带产生。第三,荧光素酶基因报告实验显示,Smad3明显增强RGC-32启动子活性,但Smad1、Smad2和Smad5对RGC-32启动子活性没有影响,故Smad3可能是TGF-β诱导的RGC-32启动子活化的关键分子。第四,将Smad2和Smad3表达质粒分别或一起与p-1500RGCluc共同转染NRK-52E细胞,Smad3明显增强RGC-32启动子活性,而Smad3 siRNA可抑制RGC32启动子活性。以上结果表明,在TGF-β诱导的NRK-52E细胞EMT过程中,Smad3可通过与RGC-32基因启动子区上游的SBE序列结合,对RGC-32的转录调控起到重要作用。

图5 TGF-β1通过Smad3调控RGC-32转录示意图TGF-β:转化生长因子β;RGC-32:补体应答基因32;SBE:Smad结合元件;TβR:转化生长因子β受体
现已明确,TGF-β/Smad信号转导通路在EMT过程中起着关键作用,Smad2和Smad3在Smad4的协助下,将TGF-β信号从细胞表面传导至细胞核,从而调控相关基因转录最终发挥其生物学作用[23]。前期工作表明RGC-32为Smads下游关键调节分子在EMT过程中起重要作用[9],但其确切分子调控机制不明。Huang等[10]研究发现RGC-32转录调控在TGF-β诱导神经嵴细胞株Monc-1细胞向平滑肌细胞分化过程中起着重要作用,且Smad2与PEA3相互作用调控RGC-32转录水平。本研究通过体外NRK-52E细胞,进一步发现TGF-β诱导EMT过程中,TGF-β通过Smad3(并非Smad2或Smad4)与RGC-32基因上游SBE序列结合,从而调控RGC-32基因转录活性。结合Guo等[11]新近研究发现TGF-β诱导人近端肾小管细胞株(HPTC细胞)EMT过程中,Smad3必须与RGC-32蛋白结合由细胞浆跨膜进入细胞核,从而调控α-SMA、ECM和E-钙黏蛋白的表达。因此,我们推测RGC-32在TGF-β诱导EMT过程中的作用机制如图5所示,即一方面TGF-β信号通过Smads转导过程中细胞质内Smad2/Smad3/Smad4形成复合物,RGC-32蛋白与Smad3结合该复合物转入细胞核内,从而发挥作用;另一方面,Smad3与RGC-32启动子区SBE结合进一步调控RGC-32转录活性。
1 Eddy AA.Molecular basis of renal fibrosis.Pediatr Nephrol,2000,15(3-4):290-301.
2 Schieppati A,Remuzzi G.Chronic renal diseases as a public health problem:epidemiology,social,and economic implications.Kidney Int Suppl,2005,(98):S7-S10.
3 Liu Y.Epithelial to mesenchymal transition in renal fibrogenesis:pathologic significance,molecular mechanism,and therapeutic intervention.J Am Soc Nephrol,2004,15(1):1-12.
4 Iwano M,Plieth D,Danoff TM,et al.Evidence that fibroblasts derive from epithelium during tissue fibrosis.J Clin Invest,2002,110(3):341-350.
5 Kalluri R,Neilson EG.Epithelial-mesenchymal transition and its implications for fibrosis.J Clin Invest,2003,112(12):1776-1784.
6 Yang J,Liu Y.Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis.Am J Pathol,2001,159(4):1465-1475.
7 Boyer B,Vallés AM,Edme N.Induction and regulation of epithelial-mesenchymal transitions.Biochem Pharmacol,2000,60(8):1091-1099.
8 Massagué J,Wotton D.Transcriptional control by the TGF-beta/Smad signaling system.EMBO J,2000,19(8):1745-1754.
9 Huang WY,Li ZG,Rus H,et al.RGC-32 mediates transforming growth factor-beta-induced epithelial-mesenchymal transition in human renal proximal tubular cells.J Biol Chem,2009,284(14):9426-9432.
10 Huang WY,Xie W,Guo X,et al.Smad2 and PEA3 cooperatively regulate transcription of response gene to complement 32 in TGF-β-induced smooth muscle cell differentiation of neural crest cells.Am J Physiol Cell Physiol,2011,301(2):C499-C506.
11 Guo X,Jose PA,Chen SY.Response gene to complement 32 interacts with Smad3 to promote epithelial-mesenchymal transition of human renal tubular cells.Am J Physiol Cell Physiol,2011,300(6):C1415-1421.
12 Sato M,Muragaki Y,Saika S,et al.Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction.J Clin Invest,2003,112(10):1486-1494.
13 Goumans MJ,Mummery C.Functional analysis of the TGF-beta receptor/Smad pathway through gene ablation in mice.Int J Dev Biol,2000,44(3):253-265.
14 Fukasawa H,Yamamoto T,Togawa A, et al.Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice.Proc Natl Acad Sci U S A,2004,101(23):8687-8692.
15 Inazaki K,Kanamaru Y,Kojima Y,et al.Smad3 deficiency attenuates renal fibrosis,inflammation,and apoptosis after unilateral ureteral obstruction.Kidney Int,2004,66(2):597-604.
16 Böttinger EP,Bitzer M.TGF-beta signaling in renal disease.J Am Soc Nephrol,2002,13(10):2600-2610.
17 Schnaper HW,Hayashida T,Hubchak SC,et al.TGF-beta signal transduction and mesangial cell fibrogenesis.Am J Physiol Renal Physiol,2003,284(2):F243-F252.
18 Lan HY,Chung AC.TGF-β/Smad signaling in kidney disease.Semin Nephrol,2012,32(3):236-243.
19 Badea T,Niculescu F,Soane L,et al.RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase.J Biol Chem,2002,277(1):502-508.
20 Fosbrink M,Cudrici C,Niculescu F,et al.Overexpression of RGC-32 in colon cancer and other tumors.Exp Mol Pathol,2005,78(2):116-122.
21 Fosbrink M,Niculescu F,Rus H.The role of c5b-9 terminal complement complex in activation of the cell cycle and transcription.Immunol Res,2005,31(1):37-46.u ML,Mauviel A.Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach.J Biol Chem,2001,276(20):17058-17062.Specificity and versatility in TGF-beta signaling through Smads.Annu Rev Cell Dev Biol,2005,21:659-693.
(本文编辑 加 则 青 松)
The molecular mechanism of TGF-β1 regulating trascription of response gene to complement 32 through Smad3 pathways
SHENYunlin,NIUXiaoling,LIUHuajie,SUNLei,KUANGXinyu,HUANGWenyan
DepartmentofNephrologyandRheumatology,ShanghaiChildren’sHospital,ShanghaiJiaoTongUniversity,Shanghai200062,ChinaCorrespondingauthor:HUANGWenyan(E-mail:hwy65@hotmail.com)
Objective:To research the molecular mechanisms of Smad3 regulation RGC-32 transcription in TGF-β1 induced Epithelial-mesenchymal Transition on NRK-52E Cells. Methodology:The NRK-52E cells were cultured and transiently transfected in vitro. Luciferase assays were performed to determine whether the SBE plays roles in TGF-β1-induced RGC-32 promoter activation. Electrophoretic mo to determine which proteins are the formations of TGF-β1-induced nuclear complex. Results:(1) The Smad biGF-β1-induced RGC-32 transcriptional activation. The SBE mutation structure significantly inhibited RGC-32 promoter activity. (2) The SBE was essential for the interaction of TGF-β1-induced nuclear proteins with RGC-32 promoter. It was found that the TGF-β-induced interaction between nuclear factors and RGC-32 promoter in EMSA, and the wild-type DNA containing SBE site as competitors abolished the formation of TGF-β-induced complex. (3) Smad2/3 was the composition of TGF-β1-inducible transcription factor complex bound to RGC-32 promoter as recognized by their specific antibodies in EMSA. (4) Smad2 and Smad3 expression plasmids were co-transfected individually into NRK-52E cells, and then Smad3 significantly increased the RGC-32 promoter activity. This suggests that Smad3, but not Smad2, be presented in the TGF-β1-inducible complex formed by nuclear proteins with RGC-32 promoter. Conclusion:These results illuminated that Smad3 may be combined with RGC-32 promoter region SBE and play an important role in transcriptional regulation in TGF-β-induced renal tubular EMT of NRK-52E cells.
response gene to complement 32 epithelial-mesenchymal transition transforming growth factor-β transcriptional regulation
国家自然科学基金资助项目(81370813);上海市科委(114119a1700);上海市卫生局优秀学科带头人项目(XBR2011010)
上海交通大学附属儿童医院肾脏风湿科(上海,200062)
黄文彦(E-mail:hwy65@hotmail.com)
2015-02-26
ⓒ 2015年版权归《肾脏病与透析肾移植杂志》编辑部所有
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:50
中日友好医院学报(2021年1期)2021-04-14 01:58:32
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
山东医药(2020年9期)2020-05-20 01:12:16
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
中国医药导报(2015年27期)2015-02-28 22:08:01
西南国防医药(2015年11期)2015-02-28 19:38:46
