
AP聚类算法求解植入(l,d)模体识别问题
2015-05-15 01:53:08张小骏
郑州大学学报(工学版) 2015年3期
陈 昆,张小骏
(西安电子科技大学计算机系,陕西西安710071)
AP聚类算法求解植入(l,d)模体识别问题
陈 昆,张小骏
(西安电子科技大学计算机系,陕西西安710071)
模体识别是运用计算机算法寻找一系列功能相近且形式相似的DNA序列片段,从而找出生物信息学中控制基因表达调控机制的转录因子结合位氛,将这种问题转化为AP聚类算法可处理的模型,然后用AP聚类得到稳定的候选模体聚类,最终利用贪心算法对问题进行求精,得出一组候选模体集,利用相对熵测度对候选模体集合进行评价并且择优输出,从而构造出一种新的模体识别算法.实验结果分别从模拟数据和真实数据证明了所提算法的有效性.
基因转录;模体识别;AP聚类算法
0 引言
基因表达在生物上的表现是通过对基因转录、翻译等过程形成蛋白质分子来实现.在DNA序列中可以启动和控制基因转录、翻译过程的相关结合位点,被称为启动子,这些启动子序列中的特殊位点被称作转录因子结合位点[1].模体(motif)识别被作为计算手段来定位转录因子结合位点[2].模体是一个短的DNA序列片段,通常长度是5~25个碱基对(base pairs).模体识别的难点在于模体并不是精确地出现在DNA序列中,而是以退化的形式出现.模体识别通常被形式化地定义如下[3]:
植人(l,d)模体识别问题:给定t条长为n的字符表{A,C,G,T}上的DNA序列集合S= {s1,s2,…,st},以及非负整数l和d,满足0≤d<l<n.模体发现问题就是找到一个l-mer(长为l的字符串)m,使得每条序列si中都存在一个与m至多有d个位置差异的l-mer mi.我们称lmer m是一个模体,mi是一个模体实例.
如果植人的模体是充分保守的,那么(l,d)模体发现问题就可以简化为计算l-mer在给定的序列S中出现的次数.一般而言,模体识别都是基于序列中l-mer的相似性比较.Buhler and Tompa采用了利用概率分析来估计(l,d)模体发现的求解难度[4].
现有的模体识别算法主要有:一种是精确类算法,此种算法需遍历整个样本空间找到每条模体实例,算法的时间复杂度过高,使实际运用有着较大的困难,具有代表性的是W INNOWER算法[3]和PMSP算法[5].另一种是非精确算法,此种算法构造初始模型,通过模体特性多次迭代找到局部最优解,因此时间复杂度较低,不足的是只能确定局部最优解.具有代表性的算法是MEME算法[6]、基于模式驱动的PairMotif+算法[7]、基于Gibbs采样DNA算法[8]、AP聚类等聚类算法.
笔者的思路是通过AP聚类[9]来解决模体识别问题.聚类算法按照特定标准,对各个样本进行无监督的分类,得出的各个聚类是一组相互相似的l-mer,它们是一组潜在的模体实例.
1 方法
1.1 AP聚类原理
AP聚类又称近邻传播聚类,是2007年Frey等人提出的一种无监督聚类算法[9].通过对目标数据集建立相似度矩阵,引人Responsibility和Availability两种信息传递,其中Responsibility表示从数据结点i信息传向到候选聚类中心k,意味着k点作为i点聚类中心的可能性;Availability表示从候选聚类中心k信息传向结点i,意味着i点归属于聚类中心k的可能性.初始目标数据集中每个元素作为聚类中心,围绕这两种信息关系进行迭代,每次迭代就是信息关系的进一步更新,直至稳定的聚类出现.
从理论上看AP聚类有这样的特点:①不必要求设定初始的聚类中心;②聚类中心真实存在;③在相似度矩阵确定的情况下,聚类结果稳定.所以较其他的聚类算法,此算法有着较稳定的聚类效果,适合模式识别这类大规模数据处理.
1.2 用AP聚类求解模体识别问题
使用AP算法求解模体识别问题的步骤分为三步:第一步对给定数据集构建图结构;第二步用AP算法找到稳定的聚类;第三步,对聚类结果求精得到植人(l,d)模体.
1.2.1 对目标数据集构建图结构
对给定的t条DNA序列建立模体识别问题的图模型,并定义图中顶点的相似性,从而得到相似度矩阵S.对于输人序列,每个长度为l的子序列对应于图中的一个顶点,那么,模体识别问题就转化成了在图中寻找稠密子图的问题.稠密子图中的大多数顶点间的权值都比较大,为了保证聚类时的高效的时间性能,图的规模不能太大.笔者采用如下的方式减小图的规模:遍历参考序列中的每个l-mer x,由x诱导出一个子图G,并对每一个子图G进行聚类.子图G的性质如下:
①顶点集合包含x,以及si(2≤i≤t)中所有的满足与x小于等于2d个差异的l-mer.
②来自于同一条输人序列中的两个顶点间无边相连.表示为负无穷大.
③来自于不同输人序列中的两个顶点间有边相连.如果它们间的距离k满足d<k<2d,边的权重为l-k,否则边的权重为10(l-k).计算边权重的方法突显了相似性高的l-mer在聚类中所起的作用.
1.2.2 用AP算法找到稳定的聚类
利用两个矩阵R和A迭代地计算来完成聚类,矩阵R代表着Responsibility信息传递关系,矩阵A代表着Availability信息传递关系.具体说来,矩阵R中的每个元素r(i,k)表示点xk作为点xi聚类中心的可能性,矩阵A中每个元素a(i,k)表示从点xi选择xk当作聚类中心的可能性.步骤是:
(1)初始化相似度矩阵,初始设置a(i,k)= 0,对参考度S[k][k]赋值,在模体识别应用中通常设置为中位数.
(2)计算样本点之间的Responsibility值.
r(i,k)=w(i,k)-max{a(i,k′)+w(i, k′)},1≤k′≤n,k′≠k,
(3)计算样本点之间的Availability值.
(4)迭代计算.
rnew(i,k)=λrold(i,k)+(1-λ)r(i,k);
anew(i,k)=λaold(i,k)+(1-λ)a(i,k).
(5)输出结果.
O=argmaxk{a(i,k)+r(i,k)}.
其中,第4个步骤引人的λ是阻尼系数,λ取值空间为[0.5,1),下面取λ=0.8.
AP聚类算法实现伪代码为:
A lgorithm 1 AP C luster
APCluster(S,n,k,m)
Input:matrix S
Output:a cluster
(1)iter←0
(2)λ←0.8
(3)while iter<m and exemp lar changed do
(4) for i←0 to n
(5) for k←0 to n
(6) for j←0 to n
(7) R(i,k)←R(i,k)-max{A(i, j)+S(i,j)}
(8) for i←0 to n
(9) for k←0 to n
(10) for j←0 to n
(11) A(i,k)←min{0,R(k,k)+sum (max{0,R(j,k)})}
(12) for i←0 to n
(13) for k←0 to n
(14) R(i,k)←λR(i,k)+(1-λ)R(i -1,k)
(15) A(i,k)←λA(i,k)+(1-λ)A(i -1,k)
(16) for i←0 to n
(17) put A(i,k)+R(i,k)into cluster
(18) iter←iter+1
(19)return cluster
1.2.3 对聚类结果求精得到植入(l,d)模体
对得到的聚类结果即O(它使得所有数据点到最近的类代表点的相似度之和最大)中的各个聚类进一步求精来得到最终的模体.
从子图G中通过AP算法可以得到多个聚类(cluster).得到的cluster虽然包含了一组相互相似的l-mer,但并不一定能够涵盖所有的模体实例,需要对聚类进一步求精,得出其他输人序列中的模体实例.通过算法1对得到的聚类进行贪心地扩展,使得聚类中的元素能够遍布在每条输人序列之中.
A lgorithm 2 C luster Refinem ent
Inpu t:a cluster C
Outpu t:a motif
(1)Q←the sequences that contain l-mers in C
(2)while|Q|<t do
(3) select a random sequence siin S-Q
(4) find an l-mer x in sisuch that the distance from x to the l-mers in C is smallest
(5) add x to C
(6) add sito Q
(7)get a motif m by aligning the l-mers in C
(8)return m
每个得到的聚类经过扩展之后,对齐聚类中的l-mer,之后便可以得到一个模体.
1.3 整体算法
模体识别的整体算法如下所示.图1为整体算法的流程图.
A lgorithm 3 M otif D iscovery A lgorithm
Inpu t:l,d,S={s1,s2,…,st}
Output:(l,d)motifs
(1)M←Φ
(2)select a random reference sequence sifrom S
(3)for each l-mer x in sido
(4) construct a graph induced by x
(5)call Algorithm 1 and get the cluster of the graph using AP algorithm
(6)for each obtained cluster C do
(7) if|C|>10 then
(8)refine C using Algorithm 2 and get a motif m
(9)add m to M
(10)sort themotifs in M
(11)return motifs with high score用相对嫡对模体进行打分排序[10]:
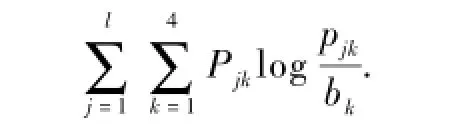
式中:pjk是输人序列中所有模体出现的第j列上碱基rk∈{A,C,G,T}的观测频率;bk是碱基rk的背景频率.相对嫡通过用于测量模体的保守性,以及模体与背景分布差异程度.
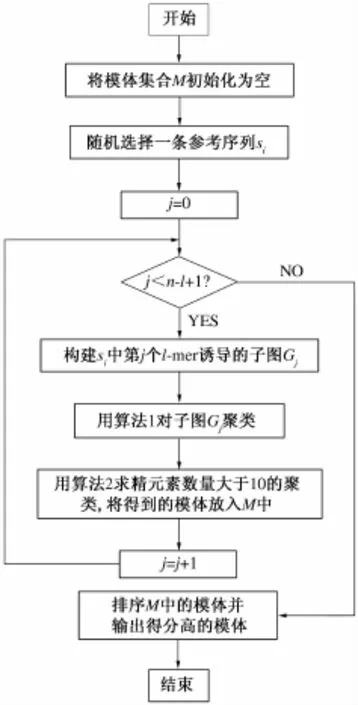
图1 整体算法的流程图Fig.1 Flow chart of the whole algorithm
2 实验与讨论
2.1 数据和评价方法
分别使用模拟数据和真实数据对所提算法进行测试.对于模拟数据,采用文献[3]中提供的标准生成方法:随机生成t条独立同分布的DNA序列和一个长为l的模体m;在每条DNA序列中的一个随机位置上植人一个模体m的实例m′,使得m′和m的最大距离为d.
对于真实数据,选用了preproinsulin、DHFR、c-fos、metallothionein和Yeast ECB5等5组生物基因序列.
用C++实现了所提算法,并与比较算法在相同的实验环境下进行了测试:惠普工作站,CPU为英特尔至强(W3505)双核2.53 GHz,内存4GB.模拟数据的所得结果是在五组随机数据上计算结果的平均值.采用核苷酸水平的性能系数(NPC)来评估识别准确率[11]:

式中:nTP表示预测的位点中所包含的已知位点的数量;nFP表示预测的位点中不是已知位点的数量;nFN表示已知的位点中没有被预测出的位点数量;nPC在准确性测量中可以同时体现特异性和敏感性.NPC的取值范围是0到1,取值越高,准确率越高.
2.2 模拟数据集上的结果
选取了3个典型的算法与所提算法进行比较,即A lignACE、MotifCut和VINE.分别是基于Gibbs采样的统计方法、基于图聚类的算法和启发式方法.
首先,通过植人不同的(l,d)实例来对算法进行比较.表1为各个算法的预测准确率.在4个比较算法中,相比AlignACE和VINE,所提算法和MotifCut都利用了全局信息,但采用了不同的聚类求精方法.在7组数据集中,所提算法有4组结果优于MotifCut,因此,在现实中所提算法的结果可以与MotifCut的结果进行相互补充.
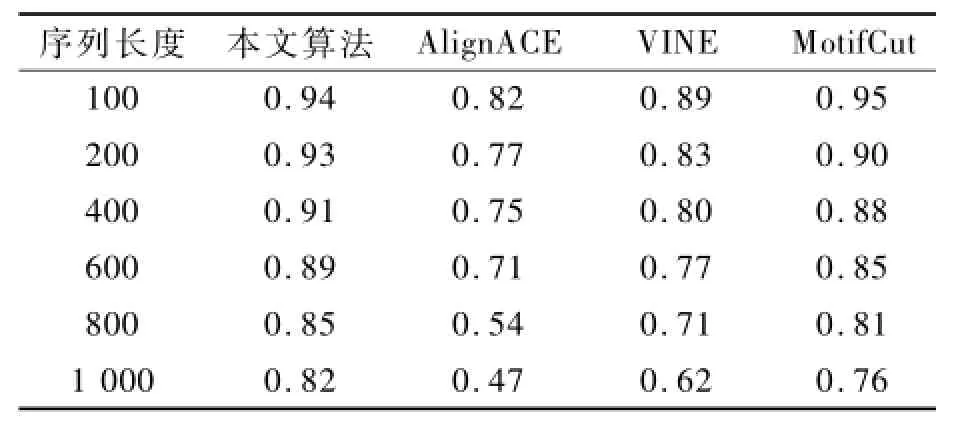
表1 不同(l,d)问题实例上的预测准确率Tab.1 Identification accuracy on different(l,d) problem instances
其次,固定(l,d)问题实例为(15,4),表2为不同序列长度的预测准确率.可以发现,所提算法的预测准确率随着序列长度的增加呈现出一个较缓的降低趋势,表明所提算法对不同的序列长度具有较好的适应性.
2.3 真实数据集上的结果
本节验证本文算法在真实数据集中识别模体的有效性.一般而言,真实数据集与模拟数据集有较大的区别.真实数据集没有模拟数据集规整,模体的保守性没有模拟数据集强,从而进一步增加了模体识别的难度.取输出结果中得分最高的模体为检测出的模体,如果检测出的模体所对应的位点能够与已知的位点有部分重叠,则认为是有效的检测;也即如果预测准确率(NPC值)大于0,则认为是有效的检测.

表2 不同序列长度上的预测准确率Tab.2 Identification accuracy on different sequence lengths

表3 真实数据集上的结果Tab.3 Results on real data sets
图2为检测出模体的序列logo图,它形象地展示出了模体的序列保守性.可以发现,检测出的模体与公布的模体有较好的相似性.更为具体的,表3给出了检测每组数据集所使用的(l,d)、得到的模体以及预测准确率.其中,检测出的模体中下划线部分表示了与公布模体重叠的部分.

图2 模体的序列logo图Fig.2 Sequence logos of detected motifs
从表3可以看出,对于Yest ECB数据,识别准确率达到了最高,主要原因是其中模体的保守性很强.对于c-fos数据,识别准确率最低,主要原因是所采用的(9,2)实例对应的序列保守性低,受到背景序列的干扰强.对于DHFH数据,虽然检测出的模体与公布的模体能够完全重叠,但是识别准确率仍然较低,原因是没有识别出所有的结合位点.
3 结论
根据算法原理和实验数据可以发现,所提AP聚类算法应用于模体识别对模体比较保守且生物背景对模体干扰较小时会取得比较好的效果.笔者虽然提出了用AP聚类算法解决模体识别的一种方法,但实际运用中AP聚类算法时间复杂度较高为O(mkN3),而且只完全适用于OOPS类型的DNA序列,对于ZOOPS类型部分适用.对于TCM类型基本不适用.若使之也可以对后两种类型适用,则m、k、N的值将更大,时间复杂度更高,所以,所提算法的改进方向是着重对初始的相似度矩阵降维修剪.
[1] HAESELEER P D.How does DNA sequencemotif discovery work[J].Nature Biotechnology,2006,24 (8),68-74.
[2] ZAMBELLI F,PESOLE G.Motif discovery and transcription factor binding sites before and after the nextgeneration sequencing era[J].Briefings in Bioinformatics,2013,14(2):225-237.
[3] PEVZNER PA,SZE SH.Combinatorial approaches to finding subtle signals in DNA sequences[C]//Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology.California: Spring-verlag,2000:269-278.
[4] BUHLER J,TOMPA M.Finding motifs using random projections[J].Journal of Computational Biology, 2002,9(2):225-242.
[5] DAVILA J,BALLA S,RAJASEKARAN S.Space and time efficient algorithms for planted motif search[C]//Proceedings of the Second International W orkshop on Bioinformatics Research and App lications.[s.l.]:CRC Press Inc,UK.2006:822-829.
[6] BAILEY T L,ELKAN C.Fitting a mixture model by expectation maximization to discover motifs in biopolymers[C]//Proceedings of the 2nd International Conference on Intelligent Systems for Molecular Biology. Berlin:Spring-verlag,1994:28-36.
[7] YU Qiang,HUO Hong-wei,ZHANG Yi,et al.Pair-Motif+:a fast and effective algorithm for De Novo motif discovery in DNA sequences[J].International Journal of Biological Sciences,2013,9(4):412 -424.
[8] LAWRENCE C E,ALTSCHUL S F,BOGUSKIM S, et al.Detecting subtle sequence signals:a Gibb's sampling strategy formultiple alignment[J].Science, 1993,262:208-214.
[9] SBRENDAN J,FERY,DELBERT D.Clustering by passingmessages between data points[J].SCIENCE, 2007,2(3):315-328
[10]GIULIO P,GIANCARLO M,GRAZIANO P.An algorithm for finding signals of unknown lengthinDNAsequences[J].Bioinformatics,2001,4(3):207-214.
[11]TOMPA M,LI N,BAILEY T L,et al.Assessing computational tools for the discovery of transcription factor binding sites[J].Nat Biotechnol,2005,23 (1):137-149.
AP Clustering Algorithm Solving Planted(L,d)Motif Identification
CHEN Kun,ZHANG Xiao-jun
(School of Computer Science,Xidian University,Xi′an 710071,China)
Transcription factors can be combined with the special DNA sequence that can control gene transcription process.The special DNA sequence is called the motifs.The motif identification is to find a set of DNA fragments with both similar functions and similar forms.It plays a crucial role in the research on the structure and function of genes.The problem was converted to themodelwhich can be processed by AP clustering algorithm.Then we get steady candidate motifs by using AP clustering.Finally we use the greedy algorithm to refine the clustering results.We can get a group of candidatemotifs set,evaluate candidatemotifs set by information content and output the optimalmotif set.Thereby the new algorithm is designed for the problem.The experimental results on both simulated data and real data demonstrate the validity of the proposed algorithm.
gene transcription;motif identification;AP clustering algorithm
TP39
A
10.3969/j.issn.1671-6833.2015.03.024
1671-6833(2015)03-0110-05
2015-01-10;
2015-03-21
中央高校基本科研项目(K50513100011)
陈昆(1975-),河南郑州人,西安电子科技大学博士研究生,高级工程师,研究方向为生物信息学,E-mail:553058474@qq.com.
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
计算机与现代化(2021年5期)2021-05-27 06:51:34
生物学通报(2019年3期)2019-02-17 18:03:58
智能计算机与应用(2019年1期)2019-01-11 06:03:06
复杂系统与复杂性科学(2017年4期)2017-07-07 02:07:48
自动化学报(2016年5期)2016-04-16 03:38:40
高中生学习·高三版(2014年3期)2014-04-29 06:11:18
高中生学习·高三版(2014年3期)2014-04-29 06:10:49
赤峰学院学报·自然科学版(2013年4期)2013-07-31 22:01:40
