
丙烷脱氢反应过程的研究Ⅱ. 石英空管反应器中氧的影响
2015-05-14 09:26:52林少波曹冬冬方锋猛隋志军朱贻安
石油化工 2015年7期
林少波,曹冬冬,方锋猛,隋志军,朱贻安,李 平
(华东理工大学 化学工程联合国家重点实验室,上海 200237)
丙烷催化直接脱氢制丙烯的工艺目前已工业化,但由于存在能耗高和催化剂易结焦失活等问题[1-2],使得人们开始研发脱氢氧化等新型催化工艺[3-4],如德国Thyssen Krupp公司已开发出UhdeSTAR催化脱氢氧化工艺[5]。
在丙烷脱氢过程中加入氧气,不仅可通过消耗氢来推动反应平衡正向移动,而且可通过氧化反应为过程提供热量,同时生成的水汽还能减缓催化剂结焦,因此对于降低反应温度和延长催化剂寿命以及节能降耗均有利。此外,现有催化剂上丙烷临氧脱氢反应温度虽较直接脱氢时下降,但仍需550℃以上才能显示出较高的活性[6-8]。高温含氧气氛下,原料丙烷和产物丙烯不但会在催化剂床层上发生深度氧化等非目标反应,而且在反应器壁上及气相空间内也有可能经自由基路径发生均相氧化反应,从而对丙烯选择性造成直接影响[9-11]。同时,氧存在下自由基引起的爆炸反应一直是低碳烃类临氧反应工艺应用时面临的潜在危险[3]。
低碳烃类的均相氧化反应在20世纪中期已有大量研究,但当时的主要目的并非烯烃的生产[11-13]。随着丙烷催化临氧脱氢制丙烯技术的兴起,人们对于该过程中烃类发生均相氧化反应的程度有诸多推测[3],有研究者甚至认为催化剂的作用只是促进了均相自由基反应[9]。迄今为止,对于丙烷脱氢体系中的均相氧化反应过程进行系统研究的报道很少。
本工作在前期研究的基础上[14],考察了钝化的石英空管反应器中,氧对丙烷和丙烯热解的影响,以期发现氧加入量与丙烷和丙烯的转化率及产物分布之间的关联性。
1 实验部分
采用磷酸钝化的石英管反应器(内径8 mm、外径10 mm、长400 mm),反应器加热炉长度为240 mm[14]。
反应原料为丙烷(或丙烯),含量为4%(x),其余为氮气。氧加入量与丙烷(或丙烯)的摩尔比为1∶8,1∶4,1∶2,1∶1,气体总流量120 mL/min,常压,温度350~700 ℃,反应管等温区内气体停留时间2.5 s。
离开反应管后的气体经冰水冷凝和硅胶干燥后采用气相色谱仪(GC9800型,上海科创仪器有限公司)进行在线分析,选用TDX-01和PQ填充柱分离H2,O2,CO,CO2,TCD检测;GDX-01填充柱分离丙烷、丙烯、乙烷、乙烯、甲烷及含氧有机物,FID检测。所有气体成分均采用外标法定量,反应尾气总量用皂膜流量计测定。
原料转化率(X)、丙烯选择性(S)和收率(Y)定义为:

式中,Fi为进口物料流量,mL/min;Fe为出口物料流量,mL/min;xi为原料进口摩尔分数;xe为原料出口摩尔分数;xeo为丙烯出口摩尔分数;xia为丙烷进口摩尔分数;xea为丙烷出口摩尔分数。
2 结果与讨论
2.1 氧对丙烷热解的影响
丙烷热解时,不同氧加入量下丙烷和氧的转化率随温度的变化见图1。

图1 不同氧加入量下丙烷和氧的转化率随温度的变化Fig.1 Effects of temperature on the conversions of C3H8 and O2 with different addition of O2.
由图1(a)可见,与无氧条件下的丙烷热解相比,有氧条件下丙烷发生反应的起始温度从600℃降至550 ℃,且氧加入量越多,相同温度下的丙烷转化率越高,如700 ℃时,无氧条件下丙烷转化率为14.0%,而当n(O2)∶n(丙烷)=1∶1时丙烷转化率可达79.5%。显然,氧的加入不但可降低丙烷的起始转化温度,还能显著促进丙烷的转化。由图1(b)可见,在600 ℃以下,氧转化率受氧加入量的影响不明显;但随温度的提高,高氧加入量体系中的氧转化率急剧上升,说明反应过程对氧浓度的依赖性逐渐增强,一些需多分子氧参与的反应(如深度氧化反应)开始发生[10]。虽然原料中氧的加入量并不高于丙烷,但同一反应条件下氧转化率始终低于丙烷转化率,说明两者的反应是非等分子数进行或反应路径不完全一致,丙烷与氧反应时的相对分子数更多或有部分丙烷发生了无氧反应。因此,在实验条件下丙烷的浓度效应更明显。
不同氧加入量下丙烷热解产物组成随温度的变化见图2。

图2 不同氧加入量下丙烷热解产物组成随温度的变化Fig.2 Product distributions in the C3H8 pyrolysis with different addition of O2.
丙烷氧化裂解的主要产物有:丙烯、甲烷、乙烯、乙烷、H2、CO。由图2可见,温度为700 ℃时,当进料为丙烷单组分时,其产物含量高低的顺序为:H2>丙烯≈甲烷>乙烯>>乙烷;当n(O2)∶n(丙烷)=1∶8时,产物含量高低的顺序为:H2>丙烯≈甲烷>乙烯>CO>>乙烷;而当n(O2)∶n(丙烷)=1∶1时,产物含量高低的顺序为:CO>H2≈甲烷≈乙烯>丙烯>乙烷。
无氧与低氧条件下的产物分布比较相似,产物中H2居多,甲烷等其次。但随氧加入量的提高,CO含量逐渐增加,并成为最主要产物。这是由于随氧加入量的增加,多分子氧参与的丙烷深度氧化反应成为了主导反应。由图2(a)可看出,当温度低于650 ℃时,丙烯的生成量随氧加入量的增加而提高,表明氧的存在也能促进丙烯的生成;但当温度升高后,高氧加入量(n(O2)∶n(丙烷)=1∶1)下产物中丙烯的含量呈下降趋势,而此时CO大量生成,表明丙烯可能被深度氧化而消耗,即丙烷氧化裂解和深度氧化反应在与丙烷氧化脱氢生成丙烯反应的竞争中占优势。从图2(b)~(d)可看出,其余的烃类产物随氧加入量和温度的提高仍呈增长趋势,未明显受到CO生成反应竞争的影响。值得一提的是,在有氧反应条件下,H2生成量一直很大,其含量始终高于烃类产物的含量,即使在高温(700 ℃)和高氧加入量(n(O2)∶n(丙烷)=1∶1)下,H2生成量仍未减少;而且丙烷反应体系中深度氧化的主要产物为CO,而非CO2(未检测到),特别是在高温和高氧加入量下,CO的生成量远超过其他产物。这些情况与热力学计算的结果[10]极其相似,说明石英空管反应器中所发生的有氧反应,在高温时基本上沿着热力学能量路径进行,遵循气相自由基反应机制。
许多研究认为,无氧条件下,丙烷气相热解第一步是C—C键断链产生自由基,随后发生自由基转移,逐步生成甲烷、乙烯、丙烯和H2,见式(1)~(4)[14]。而在有氧气氛中,丙烷气相热解的链引发步骤为式(5),同时形成丙基和活泼氧自由基物种(如HO2·),其活化能约为192.5 kJ/mol,比丙烷无氧热解(见式(1))的活化能(366.1 kJ/mol)低,因而可在更低温度下发生[15]。丙烯的生成可通过丙基自由基与氧分子直接碰撞产生(见式(6))。相比于式(4)的丙基解离形成丙烯步骤,式(6)的反应速率快得多[13,16],所以氧存在下丙烯更易形成,氧浓度越高,对丙烯生成越有利。丙基自由基的大量存在也导致了甲烷和乙烯的进一步生成,其生成路径可通过式(3)或通过丙烯氧化路径。而高温高氧量下CO的爆发式增长,很可能是由丙基通过其氧化裂解产生的含氧有机物自由基的快速传递和解离(见式(7)~(15))所引起,期间伴随更多H·(见式(14))和H2(见式(15))的产生。因此,加入氧量越多,CO生成量就越多,H2生成量也越多。由于体系中存在着大量H·,它与活泼氧自由基(HO·,O·)的反应速率显然快于CO的进一步氧化[17],同时,热力学上也不利于CO2的稳定存在[10],所以CO2的生成量极少。此外,实验中也确实检测到了少量含氧有机物成分。


图3为不同氧加入量下丙烯选择性和收率随温度的变化。由图3(a)可看出,随温度的提高,含氧气氛中的丙烯选择性先升后降,约600 ℃时达到极值,且n(O2)∶n(丙烷)=1∶8时的丙烯选择性最高(为47.7%),n(O2)∶n(丙烷)=1∶1时其次。600 ℃时有氧条件下的丙烯选择性均高于无氧条件下的丙烯选择性;但继续提高温度,有氧条件下的丙烯选择性均下降,其中,n(O2)∶n(丙烷)=1∶1时的丙烯选择性下降最快,至700 ℃时已显著低于其他进料条件下的丙烯选择性。而在无氧条件下,700 ℃之前丙烯选择性一直呈上升趋势,700 ℃时其值超过了n(O2)∶n(丙烷)=1∶8时的丙烯选择性,达37.7%。由此可见,700 ℃时丙烯选择性高低的顺序与氧加入量直接相关,氧加入量越多,丙烯选择性越低。上述结果说明,氧的加入在适宜的温度范围内对于提高丙烯选择性有利,且不同的氧加入量下其温度范围有所差异,氧加入量越少,其温度范围越宽;在适宜的温度范围内,相对更低的氧加入量可获得更高的丙烯选择性。值得注意的是,与无氧条件下相比,有氧条件下丙烯选择性高时的温度较低,表明氧的加入不但提高了丙烯的选择性,还降低了生成丙烯反应的活化能,从而使丙烯的生成反应可在较低温度下进行。
从图3(b)可看出,除了高温(700 ℃)和高氧加入量(n(O2)∶n(丙烷)=1∶1)的情况外,其余条件下的丙烯收率均随温度的上升而提高,且氧加入量越多,丙烯收率越高。但高氧加入量(n(O2)∶n(丙烷)=1∶1)时,在650 ℃附近丙烯收率出现了极大值(14.7%),至700 ℃时丙烯收率已低于n(O2)∶n(丙烷)=1∶2下的丙烯收率。这些结果也进一步体现了氧的促进作用,同时还说明适当的氧加入量及适宜的反应温度对于获得高丙烯收率具有重要意义。此外,在石英空管反应器中,实验条件下得到的最高丙烯选择性(47.7%)和收率(14.7%)与最近报道的催化剂上丙烷氧化脱氢反应的结果[18]相当,说明在这些条件下,催化剂对提高丙烯选择性和收率的作用十分有限。
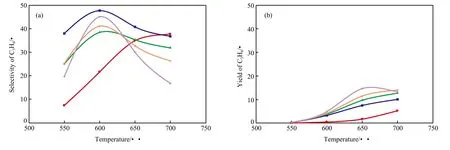
图3 不同氧加入量下丙烯选择性和收率随温度的变化Fig.3 Effects of temperature on the selectivity and yield of C3H6 with different addition of O2.
2.2 氧对丙烯热解的影响
丙烯热解时,不同氧加入量下丙烯和氧的转化率随温度的变化见图4。
由图4(a)可知,随温度和氧加入量的提高,丙烯的转化率逐步提高;但与丙烷氧化热解时的高转化率形成鲜明对比,丙烯氧化热解的程度要低得多,在高温(700 ℃)和高氧加入量(n(O2)∶n(丙烯)=1∶1)时,丙烯的转化率也只有6.0%。因此,氧的加入对于丙烯热解的促进作用并不显著,丙烯转化率对于氧含量的变化也不敏感,表明即使在含氧气氛中,丙烯也具有较高的稳定性。
由图4(b)可看出,氧的转化情况也与丙烷氧化热解过程不同,一方面,相同温度下丙烯体系中的氧转化率低了许多;另一方面,低氧加入量(n(O2)∶n(丙烯)=1∶8)下的氧转化率较高,而随氧加入量的增加,氧转化率变化不明显,表明反应体系对氧浓度的依赖性不强。但高温和高氧加入量(700 ℃、n(O2)∶n(丙烯)=1∶1)下的氧转化率有快速增长的迹象,预示着氧的浓度效应开始增强,多分子氧参与的深度氧化反应会增强。
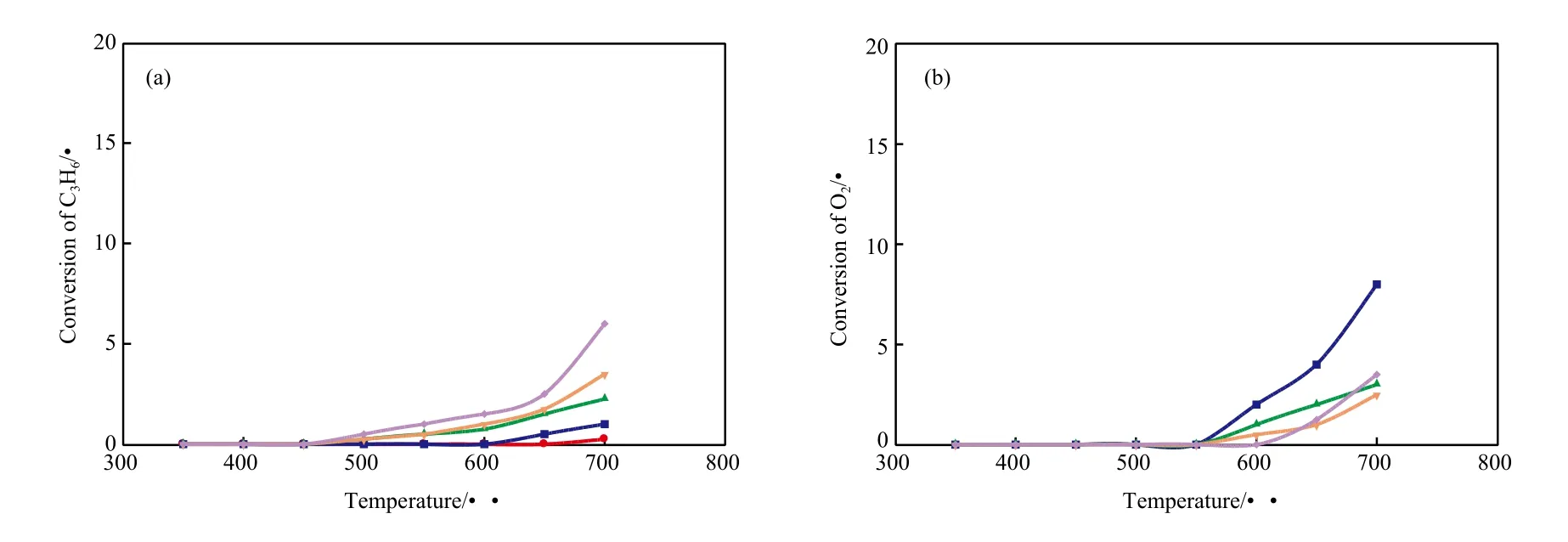
图4 不同氧加入量下丙烯和氧的转化率随温度的变化Fig.4 Effects of temperature on the conversions of C3H6 and O2 with different addition of O2.
不同氧加入量下丙烯热解产物组成随温度的变化见图5。由图5可看出,除乙烯、甲烷和H2外,在高温高氧加入量下,CO是最主要的产物。这些产物的生成量均随温度和氧加入量的提高而增加,但均远低于丙烷氧化热解的结果,如在700 ℃、n(O2)∶n(丙烯)=1∶1的条件下,CO的含量仅为相应条件下丙烷氧化热解体系的1/20。这也说明在丙烷氧化 热解体系中,丙烯并非是生成CO的主要来源。

图5 不同氧加入量下丙烯热解产物组成随温度的变化Fig.5 Product distributions in the C3H6 pyrolysis with different addition of O2.
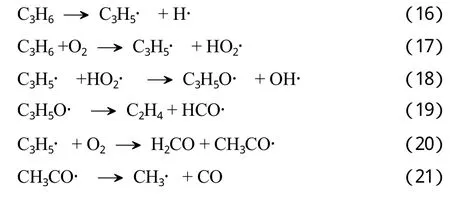
式(16)~(21)给出了丙烯脱氢和氧化的可能基元步骤。丙烯氧化的第一步与丙烯直接脱氢一样,也是生成烯丙基自由基(式(16)和式(17)),但烯丙基自由基因双键共轭效应具有较高的稳定性,造成式(18)和式(20)的链传递过程不能快速进行,因此氧气氛中丙烯也较难转化。通过式(19)、式(21)及式(14)、式(15)等步骤,烯丙基自由基可进一步通过含氧自由基中间体形成乙烯、甲烷、H2和CO等产物。随氧加入量的增多,式(20)等氧化反应将明显增强,导致CO生成量增长较快。这些实验结果与相关文献报道的结果[19-20]较一致。
3 结论
1)在钝化的石英空管反应器中,丙烷在气相中发生的均相反应过程遵循自由基机理。氧对丙烷的热解具有明显的促进作用,不仅降低了丙烷的起始反应温度,同时丙烷和氧的转化率及其产物的生成量均随原料中氧加入量的增加而增大,在650 ℃以上尤为明显。
2)不同温度下,氧加入量对丙烷氧化热解产物的分布具有不同的影响。较低温度及低氧条件下的产物分布与无氧情况相似,产物中H2居多;高温和高氧加入量下,CO是最主要的产物。氧的加入对于提高丙烯选择性和收率有利。在n(O2)∶n(丙烷)=1∶8、600 ℃和n(O2)∶n(丙烷)=1∶1、650℃的条件下,丙烯的选择性和收率达到最大值,分别为47.7%和14.7%。
3)含氧气氛中丙烯仍较难转化,氧的加入对于丙烯热解的促进作用不强。在高温高氧加入量下,CO是最主要的产物,但其生成量远低于相应条件下丙烷氧化热解体系的结果。
符 号 说 明
Fe出口流量,mL/min
Fi进口流量,mL/min
S 丙烯选择性
X 原料转化率
xe原料出口摩尔分数
xi原料进口摩尔分数
xeo丙烯出口摩尔分数
xio丙烯进口摩尔分数
xea丙烷出口摩尔分数
xia丙烷进口摩尔分数
Y 丙烯收率
[1]陈硕,王定博,吉媛媛,等. 丙烯为目的产物的技术进展[J]. 石油化工,2011,40(2):217 - 224.
[2]刘乔,董秀芹,余英哲,等. 丙烷无氧脱氢制丙烯工艺和催化剂的研究进展[J]. 石油化工,2014,43(6):713 - 720.
[3]Bhasin M M,McCain J H,Vora B V,et al. Dehydrogenation and Oxydehydrogenation of Paraff i ns to Olef i ns[J]. Appl Catal,A,2001,221(1/2):397 - 419.
[4]Czuprat O,Werth S,Caro J. Oxidative Dehydrogenation of Propane in A Perovskite Membrane Reactor with Multi-Step Oxygen Insertion[J]. AIChE J,2010,56(9):2390 - 2396.
[5]The Thyssenkrupp Company. The Uhde STAR Process:Oxydehydrogenation of Light Paraff i ns to Olef i ns[N/OL]. Thyssenkrupp-Industrial-Solutions,1999 - 12 - 01(1)[2015 - 03 -25]. http://www. thyssenkrupp-industrial-solutions.com/en/search/ searchresults.html?q=uhde+star+process&btnG.
[6]王海南,王虹. 丙烷氧化脱氢制丙烯催化剂研究进展[J]. 天然气化工,2003,28(6):25 - 31.
[7]刘永梅,曹勇,戴维林,等. Ce-Ni-O纳米复合氧化物催化剂上低温丙烷氧化脱氢[J]. 石油化工,2004,33(增刊):368 - 370.
[8]隋志军,张文军,周静红,等. 纳米碳纤维催化丙烷氧化脱氢性能的研究[J]. 石油化工,2005,34(7):612 - 616.
[9]Burch R,Crabb E M. Homogeneous and Heterogeneous Contributions to the Oxidative Dehydrogenation of Propane on Oxide Catalysts[J]. Appl Catal,A,1993,100(1):111 - 130.
[10]林少波,单玉领,隋志军,等. 氧对丙烷脱氢反应体系影响的热力学分析[J]. 化工进展,2015,34(4):1 - 7.
[11]Satterfield C N,Reid R C. Effects of Surfaces on Products Formed in the Oxidation of Propane[J]. J Chem Eng,1961,6(2):302 - 304.
[12]Jones J H,Daubert T E,Fenske M R. Oxidation and Oxidative Dehydrogenation of Ethane and Propane[J]. Ind Eng Chem Process Des Dev,1969,8(1):17 - 22.
[13]Layokun S K. Oxidative Pyrolysis of Papane[J]. Ind Eng Chem Process Des Dev,1979,18(2):241 - 245.
[14]曹冬冬,林少波,隋志军,等. 丙烷脱氢反应过程研究:Ⅰ. 空管材质和器壁的影响[J]. 石油化工,2015,44(2):18 - 26.
[15]Beretta A,Forzatti P,Ranzi E. Production of Olef i ns via Oxidative Dehydrogenation of Propane in Autothermal Conditions[J]. J Catal,1999,184(2):469 - 478.
[16]Gokulakrishnan P,Fuller C C,Klassen M S. Experiments and Modeling of Propane Combustion with Vitiation[J]. Comb Flame,2014,161(8):2038 - 2053.
[17]Mardani A,Tabejamaat S,Hassanpour S. Numerical Study of CO and CO2Formation in CH4-H2Blended Flame Under MILD Condition[J]. Comb Flame,2013,160(9):1636 -1649.
[18]贾纬华,陈明树,万惠霖. CeF3分解制备CeO2/CeF3催化剂的丙烷氧化脱氢性能[J]. 厦门大学学报:自然科学版,2014,53(4):520 - 524.
[19]Burke S M,Metcalfe W,Herbinet O. An Experimental and Modeling Study of Propene Oxidation:Part 1. Speciation Measurements in Jet-Stirred and Flow Reactors[J]. Comb Flame,2014,161(11):2765 - 2784.
[20]Xu Chaoqi,Konnov A A. Validation and Analysis of Detailed Kinetic Models for Ethylene Combustion[J]. Energy,2012,43(1):19 - 29.
猜你喜欢
扬子江诗刊(2023年3期)2023-05-06 10:40:14
大众文艺(2022年16期)2022-09-07 03:08:04
食品安全导刊(2022年2期)2022-03-18 00:35:00
食品工业(2021年12期)2021-12-31 02:57:08
天津医科大学学报(2021年4期)2021-08-21 02:14:50
农药科学与管理(2019年5期)2019-08-13 00:48:02
石油石化绿色低碳(2019年6期)2019-01-14 01:16:16
中国医药导报(2018年36期)2018-03-04 06:50:42
当代化工研究(2016年7期)2016-03-20 16:21:55
设备管理与维修(2016年6期)2016-03-16 02:22:06
