
锂取代乙烯酮与水分子之间相互作用的从头算
2015-05-08 00:55何青林刘思齐王艳红于健康
东北师大学报(自然科学版) 2015年4期
何青林,刘思齐,王艳红,于健康
(1.中国平煤神马能源化工集团有限责任公司,河南 平顶山 467000;2.吉林师范大学化学学院,吉林 四平 136000;3.中能平化天成环保公司,河南 平顶山 467000;4.吉林大学理论化学研究所,吉林 长春 130012)
锂取代乙烯酮与水分子之间相互作用的从头算
何青林1,刘思齐2,王艳红3,于健康4
(1.中国平煤神马能源化工集团有限责任公司,河南 平顶山 467000;2.吉林师范大学化学学院,吉林 四平 136000;3.中能平化天成环保公司,河南 平顶山 467000;4.吉林大学理论化学研究所,吉林 长春 130012)
在MP2/6-311++G**水平上,研究了5种复合物的几何结构型和成键特征,对能量、自然键轨道和分子中的原子进行了分析.结果表明:当锂取代3位置时,锂的电子性质突出,体系的偶极矩增加,锂的离子性质增强,通过体系共轭作用,增加了端基氧的电负性,氧的电负性质增强使得氧的给电子能力增强;当锂取代1和2位置时,锂与双键形成π-Li键或者锂与氧形成六元环结构.光谱分析表明:锂取代3位置时,红外光谱发生红移;当锂取代1和2位置时,由于锂的离子性很强,光谱发生蓝移.
自然键轨道理论;分子中的原子理论;量子化学
近年来,分子间的非化学键相互作用的研究越来越广泛,这种非键作用在化学、物理和生物领域十分重要.[1-2]毋庸置疑,氢键是这些非键作用中最重要的一类作用,一直受到人们的关注[3].随着研究的深入,人们发现了许多非标准含义上的氢键[4-5],如体系中存在大的共轭结构时,Π电子可以提供电子作为质子受体接受氢形成π-H键.
非键作用有很多独特的性质,由于偶极相互作用,使得很多体系偏离平衡状态.[6-7]氢键的强度取决电子给体和受体的结构,并且受配体结构的影响.[8-10]如果拉电子结构引入电子受体结构中,可以使得氢键作用增强.近来,很多学者研究了金属配体引起的氢键强弱变化[11],金属既可以直接参与氢键形成,也可以间接影响氢键作用.[12-13]
本文研究了乙烯酮和乙烯酮基锂与水分子形成复合物的性质,通过从头算方法详细了研究了5种复合物的几何构型和成键特征,并且做了相关的光谱和成键分析.
1 计算方法
全部计算采用Gaussian03程序.使用MP2方法,在6-311++G(d,p)基组的水平下,优化单体和复合物的几何结构,得到可靠的几何构型和频率.在此基础上,进行完全均衡校正法(counterpoise procedure,CP)计算,以校正基组叠加误差(BSSE).采用自然键轨道理论(NBO)和分子中的原子理论(AIM),对自然键键序及锂键结构和性质等进行了分析.
2 结果与讨论
在MP2/6-311++G**水平下,优化的乙烯酮和乙烯酮基锂的几何结构见图1,优化乙烯酮和乙烯酮基锂与水复合物的几何结构见图2.在MP2/6-311++G**水平下,计算的5种复合物未修正的相互作用能(ΔE)、经BSSE修正的相互作用能(ΔECP)和零点能(ΔEZPE)见表1,5种复合物的电荷以及二阶微扰稳定化能见表2,计算的复合物键鞍点处的电子密度拓扑性质见表3.
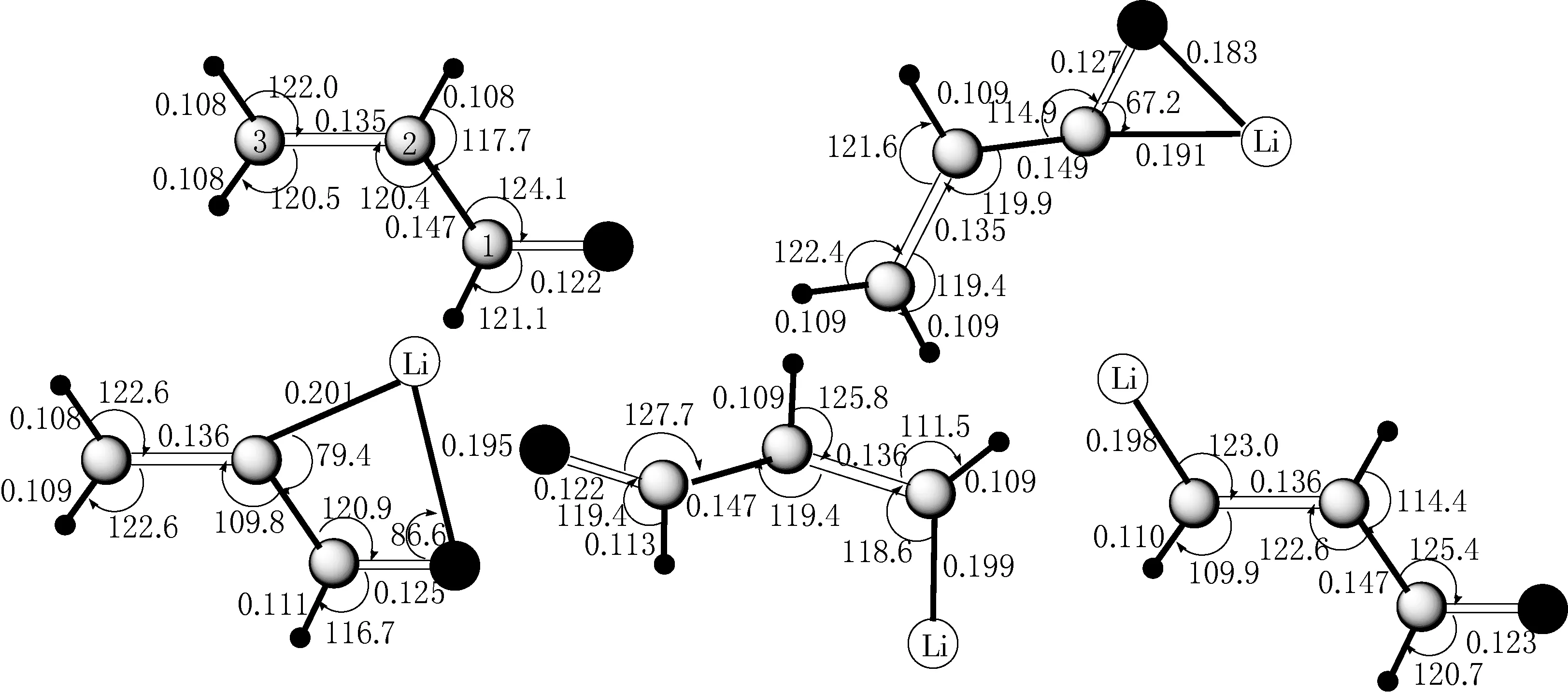
键长单位为nm;键角单位为(°)
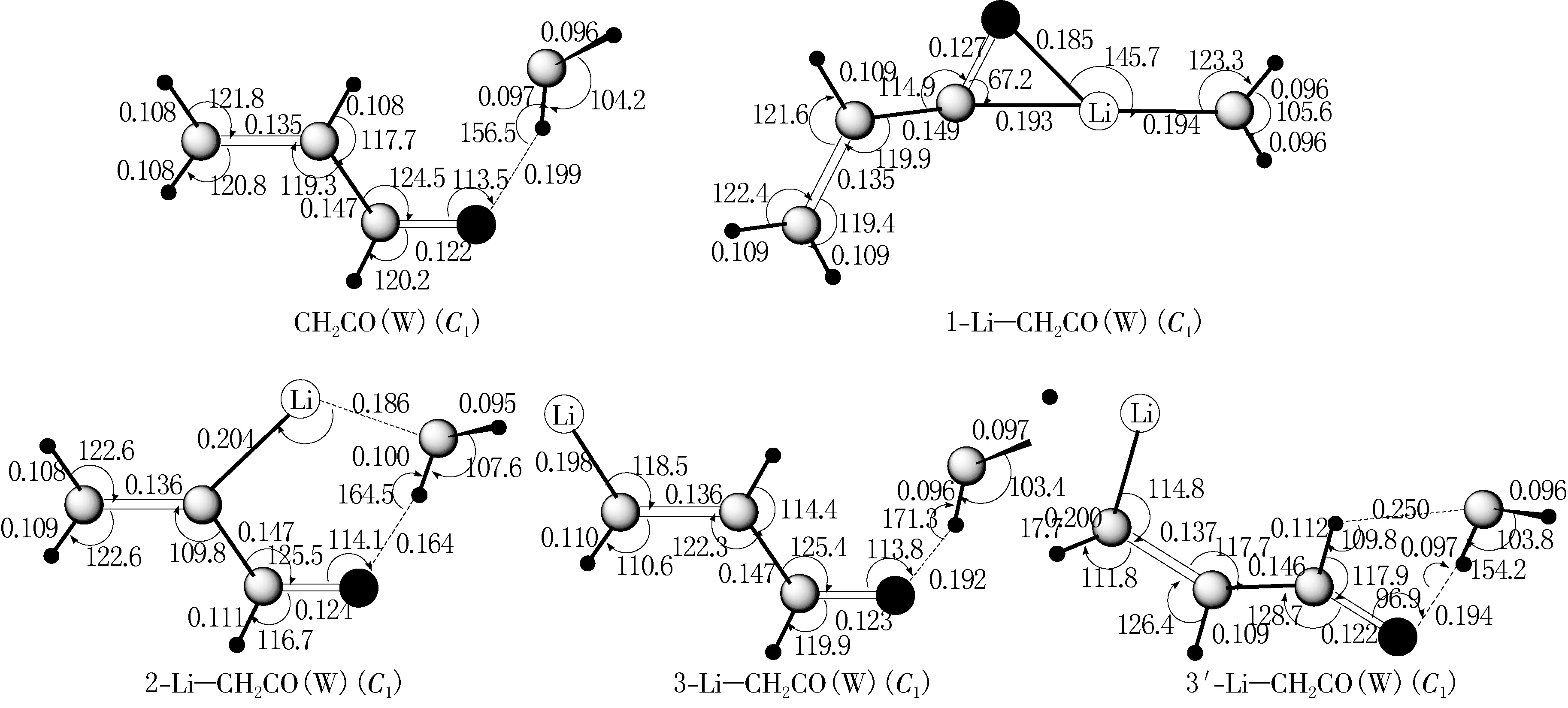
键长单位为nm;键角单位为(°)
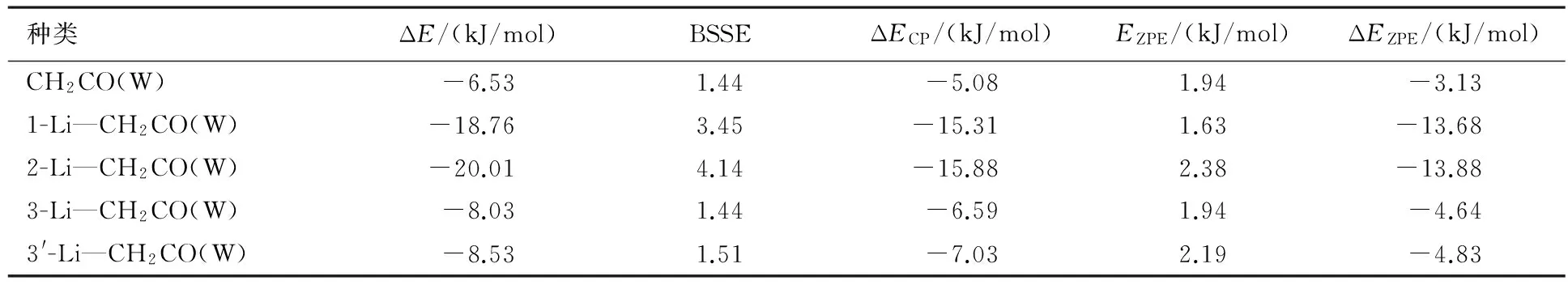
表1 在MP2/6-311++G**水平下,计算的5种复合物的ΔE,ΔECP和ΔEZPE
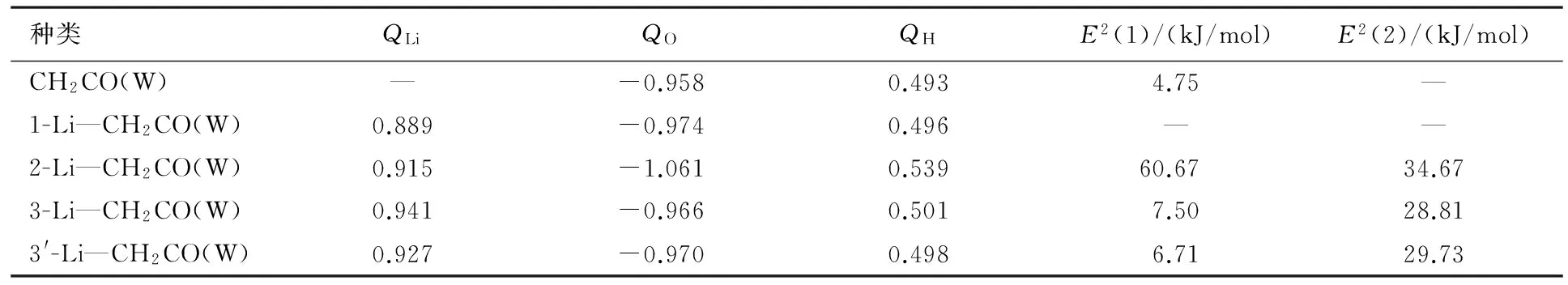
表2 在MP2/6-311++G**水平下,计算的5种复合物的电荷(Q)以及二阶微扰稳定化能(E2)
注:E2(1)和E2(2)分别来自LP(O)→σ*(O—H)和LP(C)→LP/(M)轨道相互作用.
图1表明,锂原子取代1和2位置的氢,锂与羰基相互作用,形成Li—C—O三元环和Li—C—C—O四元环,环的形成影响氢键的形成.从图2中可见:1-Li—CH2CO(W)会形成Π-Li(W)型的锂键;2-Li—CH2CO(W)中形成O—H—O氢键和C—Li—O键,2-Li—CH2CO(W)中存在弱的六元环,分子内环的形成有利于稳定分子.表1中能量表明,2-Li—CH2CO(W)在这几种复合物中能量最低.当锂取代1和2位置时,锂会与双键形成π-Li键或者锂与氧形成六元环结构,这种结构特性使得体系中氧锂键成为主要作用,与氢键相比较,体系能量更低.3-Li—CH2CO(W)和3′-Li—CH2CO(W)中锂取代3和3′位置的氢,锂的电子效应通过氢键来传递,3-Li—CH2CO(W)和3′-Li—CH2CO(W)中的氢键比CH2CO(W)中的氢键要强.从计算的偶极矩上看,当锂取代了3位置上的氢后,体系的偶极矩加大,从1.014×10-29C·m分别增加到2.96×10-29和2.11×10-29C·m,偶极矩的增加使得体系电荷分布改变,锂的取代使得乙烯酮中端基氧的负电荷增多,离子性增强.综上所述,当锂取代3位置时,锂的电子性质突出,体系的偶极矩增加,锂的离子性质增强,通过体系共轭作用,增加了端基氧的电负性,氧的电负性增强使得氧的给电子能力增强,与水分子形成的氢键作用增大.
为了研究键的性质,我们使用了AIM理论对复合物进行了分析.根据“分子中的原子”理论,电荷密度的Laplacian量2ρ是ρ的二阶导数.键鞍点处的电子密度与原子之间的成键强度有关,ρ值越大,说明该化学键的强度越大;反之,则越小.键鞍点处的2ρ与键的性质有关,一般2ρ<0,键鞍点的电荷密集,并且该值越负,化学键的共价性越强;2ρ>0,键鞍点电荷发散,并且该值越正,化学键的离子性越强;2ρ接近于0时,一般形成的是较弱的化学键.复合物的电子密度拓扑性质分析见表3,从Laplacian的值来看,体系主要以弱的共价性为主,当锂取代乙烯酮中氢后,体系的偶极增强,离子性质增大,氢键的离子性增强.
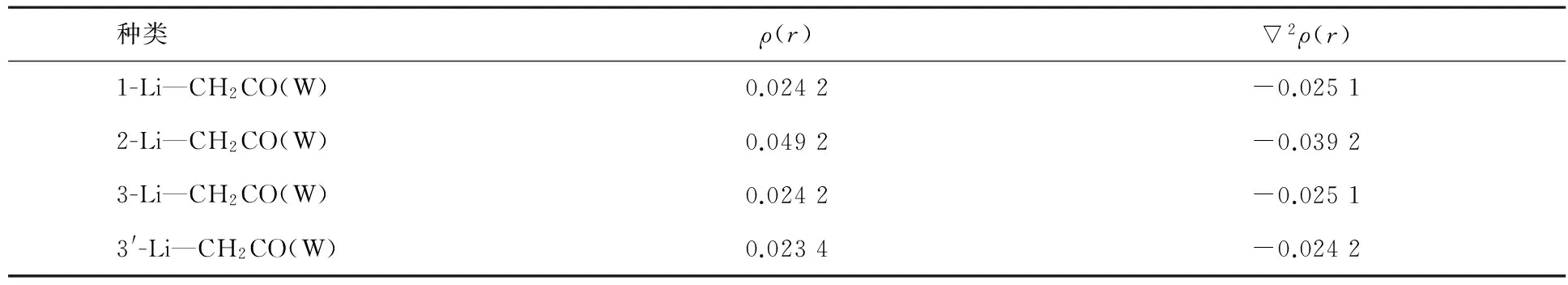
表3 复合物键鞍点处的电子密度拓扑性质
注:表中ρ(r)为电子密度,2ρ(r)为Laplacian量.
复合物中氢键键长(R)、O—H键键长(r)与水键长的差值(Δr)、O—H振动频率(ν)与水中O—H振动频率的差值(Δν)的计算值见表4.从键长看,3-Li—CH2CO(W)和3′-Li—CH2CO(W)中的O—H键键长增大了0.001 11 nm,特征振动红外光谱O—H伸缩振动红移,红移后的红外频率比水中频率小177 cm-1.2-Li—CH2CO(W)中O—H—O氢键和C—Li—O键同时存在,使得体系能量更低,红外光谱红移增大,为738 cm-1.表4中1-Li—CH2CO(W)为π-Li(W)键,水为电子给体,锂为电子受体.π-Li键长减小0.003 16 nm,频率增加了55 cm-1.这是由于水作为电子给体,减小了三元环中π-Li相互作用,使得体系中特征π-Li伸缩振动频率蓝移.这种蓝移是由于锂的离子性质在体系中占据主要作用,类似于溶剂效应中的蓝移.
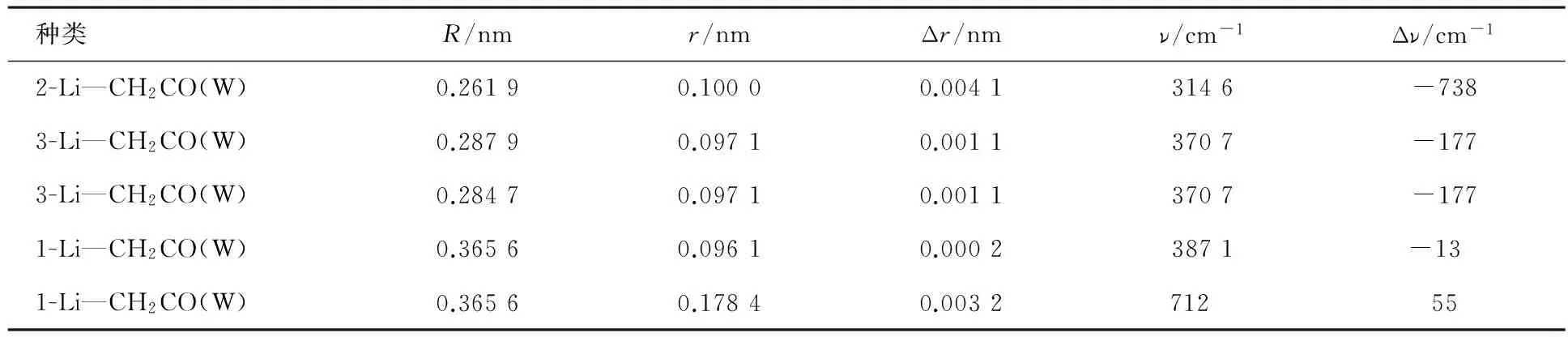
表4 复合物各种参数的计算值
3 结论
锂取代乙烯酮与水形成的氢键复合物性质有如下特征:当锂取代3位置时,锂的电子性质突出,体系的偶极矩增加,锂的离子性质增强,通过体系共轭作用,增加了端基氧的电负性,氧的电负性增强使得氧的给电子能力增强,与水分子形成的氢键作用增大,体系中氢键的离子性增强;当锂取代1和2位置时,锂与双键形成π-Li键或者锂与氧形成六元环结构,这种结构特性使得体系中锂氧键作用成为主要作用,与氢键相比较,体系能量更低.光谱分析表明,锂取代3位置时,红外光谱红移;当锂取代1和2位置时,由于锂的离子性很强,并且体系以锂键为主,光谱发生蓝移,类似于溶剂效应中的蓝移.
[1] 李刚,胡斯宪,陈琳玲.原子荧光光谱分析技术的创新与发展 [J].岩矿测试,2013,32(3):358-363.
[2] 秦超,许林.有机官能化的多金属氧酸盐研究进展[J].分子科学学报,2008,24(2):97-106.
[3] 中原武利陈鸿铿.原子荧光光谱分析[J].分析试验室 1985,4(11):34-40.
[4] 刘广平,宣益民,韩玉阁,等.一维光子晶体热辐射光谱控制模拟研究 [J].工程热物理学报,2007,28(3):475-477.
[5] 王雪莲,郭楠,李麒,等.原子荧光光谱法测定土壤样品中的硒[J].四川地质学报,2014,34(3):459-476.
[6] 舒永红.原子吸收和原子荧光光谱分析[J].分析试验室,2005,24(2):81-92.
[7] 张浩,于健康,孙家锺.从头算量子化学计算方法对高氯酸镁溶液中存在的离子缔合物种及缔合过程的研究[J].化学学报,2010,68(14):1363-1369.
[8] 张浩,于健康,孙家锺.用从头算法研究高氯酸钠溶液中存在的离子缔合物种及缔合过程[J].高等学校化学学报,2010,31(8):1600-1604.
[9] 彭琦,刘晓龙,陈佳豫.一种简易矩形波发生器设计[J].东北师大学报(自然科学版),2013,45(4):77-81.
[10] 张科,马云飞,孙乙庭,等.新型杂化材料[(2,2′-bipy)2Cu][((2,2′-bipy)2Cu)2PW12O40]·4H2O的水热合成及结构表征[J].东北师大学报(自然科学版),2014,46(3):99-104.
[11] 汪冬梅,王海水,曾广赋,等.二次微分和傅里叶解卷积技术的表观分辨率研究[J].光谱学与光谱分析,2004,24(2):152-154.
[12] 程晓丽,霍丽华,高山,等.广义二维相关光谱在红外和拉曼光谱研究中的应用[J].大学化学,2003,18(5):26-30.
[13] ZEXIAN CAO.Intermediate sp-hybridization for chemical bonds in nonplanar covalent molecules of carbon[J].Chinese Physics B,2014(6):145-149.
(责任编辑:石绍庆)
Ab initio study of the structure and vibrational properties in the Li-ketenes-water complexes
HE Qing-lin1,LIU Si-qi2,WANG Yan-hong3,YU Jian-kang4
(1.China Ping Coal Horse Energy Chemical Refco Group Ltd,Pingdingshan 467000,China;2.School of Chemistry,Jilin Normal University,Siping 136000,China;3.The Energy Level of Tiancheng Environmental Protection Company,Pingdingshan 467000,China;4.Institute of the Theoretical Chemistry,Jilin University,Changchun 130012,China)
The structures and properties of four Li-ketenes-water complexes has been fully studyed at the MP2/6-311++G(d,p) level.By studying the energy,nature bond orbital and atom-in molecules,the property of different Li-ketenes-water complexes has been discovered.When the Li at C3,the ironic of Li increased the dipole moments of Li-ketenes-water complexes.This property effects the electron density of oxygen in the ketene and the interaction between Li-ketenes and water.When the Li at C1or C2,theπ-Li bond or the six-atom-ring formed,these structure are prefer to the Li-bonding interation.The calculated vibrational frequencies of Li-ketenes-water complexes indicate that the red shifts are related to the C3,and the blue shift to the C1and C2.
natural bond orbital theory;atom-in-molecules theory;quantum chemistry
1000-1832(2015)04-0102-04
10.16163/j.cnki.22-1123/n.2015.04.022
2014-04-23
国家自然科学基金资助项目(20993149).
何青林(1967—),男,工程师,主要从事煤矿产业环境保护研究;通讯作者:于健康(1975—),男,副教授,主要从事物理化学研究.
O 64 [学科代码] 150·30
A
猜你喜欢
疯狂英语·爱英语(2020年6期)2020-07-04
疯狂英语·新策略(2020年6期)2020-06-28
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
中成药(2017年5期)2017-06-13
中学化学(2015年12期)2016-01-19
化工管理(2015年11期)2015-08-15
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年1期)2014-02-28
物理化学学报(2012年2期)2012-12-05
