
固相萃取/高效液相色谱法测定水中痕量硝磺草酮
2015-04-27 03:12王小梅谭培功曹正梅郎印海
分析测试学报 2015年2期
王小梅,谭培功 ,曹正梅,郎印海*
(1.中国海洋大学 环境科学与工程学院,山东 青岛 266100;2.中国海洋大学 海洋环境与生态教育部重点实验室,山东 青岛 266100;3.青岛市环境监测中心站,山东 青岛 266100)
固相萃取/高效液相色谱法测定水中痕量硝磺草酮
王小梅1,2,谭培功3,曹正梅3,郎印海1,2*
(1.中国海洋大学 环境科学与工程学院,山东 青岛 266100;2.中国海洋大学 海洋环境与生态教育部重点实验室,山东 青岛 266100;3.青岛市环境监测中心站,山东 青岛 266100)
建立了一种固相萃取/高效液相色谱(SPE/HPLC)测定水中痕量硝磺草酮的方法。水样用磷酸调至pH 3.0~4.0,经C18固相萃取柱富集、乙腈-水(1∶1)洗脱后,用HPLC/紫外检测器测定。测定条件为:流动相为乙腈-磷酸水溶液(45∶55,pH 3.0),流速0.5 mL/min,色谱柱为Thermo Hypersil-C18柱(250 mm×4.6 mm,5 μm),柱温30 ℃,检测波长233 nm。结果表明:在0.4~20 μg·L-1范围内,硝磺草酮的线性良好,方法检出限为0.039 μg·L-1。加标浓度为1,5,10 μg·L-1时,硝磺草酮的回收率为90.3%~94.7%,相对标准偏差为3.5%~4.6%。应用该方法测得实际水样中硝磺草酮的加标回收率为89.9%~93.2%。该方法具有快速、简便、灵敏等优点,可用于水样中痕量硝磺草酮的测定。
硝磺草酮;固相萃取;高效液相色谱;乙腈;水
硝磺草酮(Mesotrione),化学名为2-(4-甲磺酰基-2-硝基苯甲酰基)环己烷-1,3-二酮,商品名为米斯通,是一种近年来广泛使用的新三酮类除草剂[1]。硝磺草酮具有较高的起始活性、残留活性,较强的水溶性和致癌性等特点,极易对地表水和地下水造成污染[2]。硝磺草酮在迁移和转化过程中能通过光解、水解、挥发等途径减少环境中的残留量,但仍有可能通过远距离迁移进入水体,并通过食物链作用最终对生物和人体健康产生影响[3-4]。EPA于2007年报道了在1961~1990年间某一地区以硝磺草酮作为除草剂施用于农作物,使得该区域水中硝磺草酮的含量为0.1~3 μg·L-1[5]。目前,我国尚未规定水中硝磺草酮含量的限值。
水中硝磺草酮的提取方法通常采用液液萃取和固相萃取。液液萃取[6-7]方法的操作过程较繁琐,且需要消耗大量有机试剂。固相萃取的操作简便、环境污染小[8],且能有效去除样品中的杂质从而提高检测灵敏度[9],近年来被广泛用于水中有机污染物的提取。常用的硝磺草酮测定方法主要有HPLC/荧光检测法[10]、HPLC/紫外检测法[11-12]、HPLC-核磁共振[13]和 HPLC-质谱法(MS)[14]。HPLC-核磁共振和HPLC-MS可提高定量分析的选择性和灵敏度,但也会增加分析检测的成本和运行费用。Alferness等[10]采用C18柱萃取玉米、土壤和水中的硝磺草酮及其代谢物,经衍生化后利用HPLC/荧光检测器进行定量测定。Gervais等[15]利用Water Oasis HLB柱萃取水中的硝磺草酮,乙腈-二氯甲烷(1∶1)洗脱后,利用超高效液相色谱-质谱法(UPLC-MS)进行测定。 尽管国外学者开展了环境中硝磺草酮的固相萃取和测定研究[16-17],但主要集中于探讨多种污染物的固相萃取以及采用HPLC-MS测定,对于不同萃取柱、不同洗脱溶剂和洗脱体积的研究较少,国内也未见相关报道。本文探讨了不同萃取柱、不同洗脱剂、洗脱体积、上样速度和洗脱速度对萃取效率的影响,建立了C18固相萃取水中痕量硝磺草酮的方法;通过比较分析检测波长和流动相比例对分离度的影响,优化了HPLC测定条件,从而确定了水中痕量硝磺草酮测定的固相萃取/高效液相色谱方法。
1 实验部分
1.1 仪器与试剂
LC-10ATvp高效液相色谱仪(日本岛津公司);Thermo Hypersil C18柱(250 mm × 4.6 mm,5 μm,美国热电公司);Agilent Zorbax SB-C18柱(250 mm × 4.6 mm,5 μm,北京金欧亚科技发展有限公司);KQ3200B型超声波清洗器(昆山市超声仪器有限公司);ASE-24固相萃取仪(天津奥特赛恩斯仪器有限公司);玻璃纤维滤膜(0.45 μm,Whatman公司);HLB固相萃取柱(500 mg,6 mL,美国Waters公司);C18固相萃取柱(1 000 mg,6 mL,天津博纳艾杰尔科技有限公司);Multi 3430便携式多参数水质分析仪(德国WTW公司)。
乙腈(色谱纯,德国Merck公司);磷酸(分析纯,南京化学试剂有限公司);甲醇(色谱纯,瑞典Oceanpak 公司);二氯甲烷(色谱纯,天津市科密欧化学试剂有限公司);丙酮(色谱纯,北京迈瑞达科技有限公司);硝磺草酮标准品(纯度≥99.0%,德国Dr.Ehrenstorfer公司);实验用水为超纯水。
1.2 标准溶液的配制
标准品储备液:精密称取硝磺草酮标准品适量,用少量乙腈溶解后,再用水准确定容,配成浓度为100 mg·L-1的标准品储备液,于4 ℃下保存备用。标准品系列溶液:分别准确移取适量标准品储备液于容量瓶中,加水稀释,配成所需浓度的标准品系列溶液。
1.3 实际水样的采集与预处理
采集青岛某地表水样和农药厂废水置于棕色玻璃瓶中,用磷酸调至pH 3.0~4.0,于4 ℃下保存备用。水样经0.45 μm玻璃纤维滤膜过滤后,直接进行固相萃取。
1.4 固相萃取步骤
C18固相萃取柱使用前依次经10 mL乙腈-水(1∶1)和10 mL水活化,在活化过程中保持柱体湿润。取1 000 mL水样通过活化后的C18固相萃取柱,保持水样流速为3 mL/min,待水样全部过柱后,抽真空干燥3 min。再用10 mL乙腈-水(1∶1)为洗脱剂洗脱,洗脱速度为3 mL/min,洗脱后抽真空干燥3 min,收集洗脱液并用洗脱剂定容至10 mL,待测。
1.5 色谱条件
Thermo Hypersil C18柱(250 mm×4.6 mm,5 μm);高效液相色谱的检测波长为233 nm,柱温30 ℃,流动相为乙腈-磷酸水溶液(45∶55,pH 3.0),进样量为20 μL,流速为0.5 mL/min。
2 结果与讨论
2.1 固相萃取方法的优化
2.1.1 固相萃取柱的选择 考察了C18和Water Oasis HLB两种固相萃取柱对硝磺草酮的分离效果。由于硝磺草酮呈弱酸性(pKa=3.2),因此在过萃取柱前将硝磺草酮pH值调至3.0~4.0,使硝磺草酮能以分子形式与填料相结合,从而被保留在固相萃取柱中。实验结果表明,两种固相萃取柱对硝磺草酮的分离效果相似,平均回收率均大于 89.7%。考虑价格因素的差异,最终确定固相萃取柱为C18柱。
2.1.2 洗脱剂的选择 不同洗脱剂对回收率具有一定影响,为实现最佳的洗脱效率,本文分别比较了甲醇、甲醇-乙腈(1∶1)、甲醇-水(1∶1)、乙腈-水(1∶1)、二氯甲烷和丙酮6种常用溶剂对硝磺草酮的洗脱效果,洗脱体积均为10 mL。结果表明,丙酮和甲醇-水(1∶1)不能将硝磺草酮洗脱出来;经二氯甲烷洗脱后,硝磺草酮的回收率为34%;其余洗脱剂所得到的回收率均达到89.7%以上。考虑到高效液相色谱的流动相为乙腈-磷酸水溶液,为保持一致性,选择乙腈-水(1∶1)为洗脱剂。
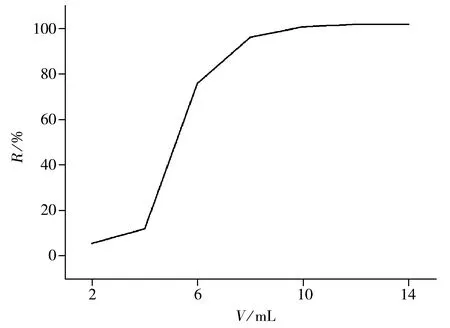
图1 洗脱剂体积对回收率的影响
2.1.3 洗脱剂的用量 在1 000 mL空白水样中加入一定浓度的硝磺草酮标准品,以乙腈-水(1∶1)为洗脱剂,分7次洗脱,每次2 mL,分段收集洗脱液并定容至10 mL,测定其回收率(见图1)。结果发现,第5次洗脱后的回收率为100.8%,且已基本稳定,因此选择洗脱剂的最佳用量为10 mL。
2.1.4 上样速度 根据文献[18-19]报道,对于大体积水样,一般选择5 mL/min和7 mL/min的上样速度。本实验在此基础上分别研究了在3,5,7 mL/min的上样速度下硝磺草酮的回收率。结果表明,在3种上样速度下,硝磺草酮的回收率均在84%以上,其中上样速度为3 mL/min时,硝磺草酮的回收率最高(93%)。因此,实验选择最佳上样速度为3 mL/min。
2.1.5 洗脱速度 选择适宜的洗脱速度,既能使目标化合物最大限度地洗脱干净,又能节省时间。本实验分别研究了1,3,5 mL/min的洗脱速度对硝磺草酮回收率的影响。结果表明,3种洗脱速度下,硝磺草酮的回收率均在88%以上,其中洗脱速度为3 mL/min时,硝磺草酮的回收率最高(93%)。因此,实验选择最佳洗脱速度为3 mL/min。
2.2 高效液相色谱条件的优化
2.2.1 检测波长的选择 为确定最佳测定波长,使用二极管阵列检测器,分别对空白及硝磺草酮进行光谱扫描,扫描范围为190 ~800 nm。结果表明,硝磺草酮的最大吸收依次出现在220,268,233 nm。在波长220 nm处,干扰过多;波长为268 nm处,方法灵敏度不高。因此确定233 nm作为紫外检测器波长。
2.2.2 色谱柱的选择 采用乙腈-磷酸水溶液(45∶55,pH 3.0)为流动相,流速为0.5 mL/min,对比Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm)和Thermo Hypersil C18柱(250 mm×4.6 mm,5 μm)对硝磺草酮分离性能的影响。实验结果表明:在上述测定条件下,Agilent Zorbax SB-C18柱和Thermo Hypersil C18柱的分离度分别为6.48与7.24,均能较好分离硝磺草酮。因此使用Thermo Hypersil C18柱可满足分析要求。
2.2.3 流动相的选择 根据国内外学者的研究,用高效液相色谱法测定硝磺草酮的流动相种类主要为甲醇-磷酸溶液[1,6]和乙腈-磷酸溶液[3,12,20-21]。本文分别比较了这两种溶液为流动相时硝磺草酮的分离情况。结果表明,以乙腈-磷酸溶液为流动相时,硝磺草酮的出峰时间为11.875 min,峰形良好,分离度为7.24。以甲醇-磷酸溶液为流动相时,硝磺草酮的出峰时间为27.5 min,出峰时间过长,峰形不佳。因此,实验选择乙腈-磷酸溶液为流动相对硝磺草酮进行分离。由于硝磺草酮为弱酸性农药(pKa=3.2),流动相的pH值对分离具有一定影响。本文比较了流动相在不同pH值(6.0,5.0,4.0和3.0) 时对硝磺草酮分离度的影响。结果表明,当pH值在4.0~6.0范围时,硝磺草酮的出峰时间早,不能与溶剂峰较明显的分离,分离效果较差。当pH值为3.0时,硝磺草酮的峰形良好、分离效果较好,分离度为7.24,因此确定乙腈-磷酸水溶液流动相的pH值为3.0。进一步分析了乙腈-磷酸溶液流动相在不同比例(30∶70,35∶65,40∶60,45∶55)时硝磺草酮的分离情况。结果表明,当乙腈-磷酸溶液比例为30∶70和35∶65时,硝磺草酮的分离度分别为9.93和14.66,但色谱峰的保留时间过长。流动相比例为40∶60和45∶55时,硝磺草酮的分离度分别为 8.17和7.24,但后者对硝磺草酮的响应值更高。故确定最佳流动相为乙腈-磷酸(45∶55,pH 3.0)。在优化条件下,1 000 μg·L-1硝磺草酮标准溶液的色谱图见图2,其峰形对称,分离效果好。
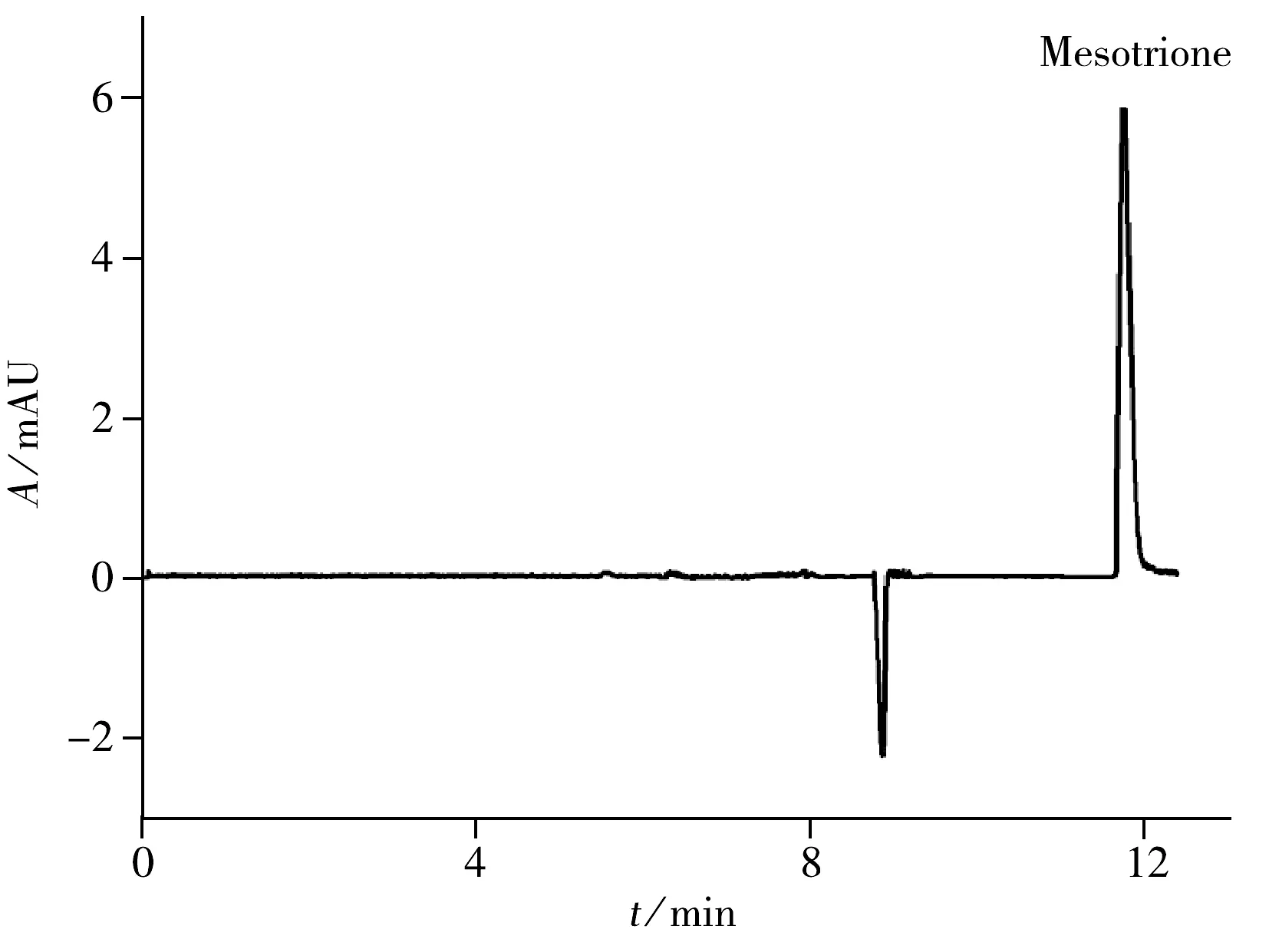
图2 1 000 μg·L-1硝磺草酮标准溶液的色谱图
2.2.4 标准曲线与方法检出限 分别配制浓度为0.4,1,5,10,20 μg·L-1的硝磺草酮系列标准溶液,按照优化色谱条件进行测定,以浓度(x,μg/L)为横坐标,对应峰面积(y)为纵坐标绘制标准曲线。结果表明,硝磺草酮浓度在0.4~20 μg·L-1范围内线性关系良好,相关系数为0.999 7。利用MDL=3.143×δ(δ为7次测定值的标准偏差)计算方法检出限。取硝磺草酮质量浓度为0.4 μg·L-1的水样,平行测定7次,当固相萃取1 000 mL水样时,方法的检出限为0.039 μg·L-1。
2.2.5 回收率与精密度 分别在空白水样中添加硝磺草酮标准品,使水样中硝磺草酮的浓度分别为1,5,10 μg·L-1,按上述前处理方法和色谱条件进行测定,每个浓度平行测6次。结果显示,3个加标水平下的回收率分别为90.3%,91.7%,94.7%,相对标准偏差(RSD) 分别为3.5%,3.5%,4.6%,方法显示了较好的回收率与精密度。
2.3 实际水样的测定
按照上述方法对地表水样和农药厂废水进行硝磺草酮含量的测定。为进一步考察该方法用于实际水样测定时的准确度,加入标准溶液进行加标实验。结果表明,地表水中未检出硝磺草酮,农药厂废水中检出硝磺草酮的浓度为1.31 μg·L-1。向地表水和农药厂废水样品中分别加入5,10 μg·L-1硝磺草酮标准溶液,连续测定6次,平均加标回收率及相对标准偏差见表1。结果表明,硝磺草酮的平均加标回收率为89.9%~93.2%,相对标准偏差(RSD)为3.6%~5.8%。实际水样中硝磺草酮的加标回收率良好,该方法可用于实际水样的检测。

表1 实际水样中硝磺草酮的加标回收率与相对标准偏差(n=6)
3 结 论
本实验采用固相萃取/高效液相色谱法测定水中痕量硝磺草酮,样品用C18固相萃取柱分离富集,以乙腈-水(1∶1)为洗脱剂,采用Thermo Hypersil-C18柱(250 mm×4.6 mm,5 μm)分离,乙腈-磷酸溶液(45∶55,pH 3.0)为流动相。该方法的检出限为0.039 μg·L-1,对实际水样的加标回收率为89.9%~93.2%。该方法具有快速、简便、灵敏度高等优点,可用于水中痕量硝磺草酮的测定。
[1] Sun Y B,Xu Y M,Sun Y,Qin X,Wang Q,Gao Y.Environ.Chem.(孙约兵,徐应明,孙扬,秦旭,王倩,高阳.环境化学),2013,32(1):144-149.
[2] Zhang D H,Teng G S,Li Z Q,Li A J,Kang M Q,Mou J.Chin.J.Anal.Chem.( 张代辉,腾国生,李正强,李爱军,康明芹,牟峻.分析化学),2012,40(5):811-812.
[3] Duan Y L,Tang B H,Huang X,Li Y,Huang C T,Cai L M.Agrochemicals(段亚玲,汤保华,黄鑫,李莹,黄成田,蔡磊明.农药),2013,52(8):582-584.
[4] Li A J,Qi B,Li F L,Mou J,Wang M T,Zhang D H,Fu R.Phys.Test.Chem.Anal.:Chem.Anal.( 李爱军,齐犇,李凤兰,牟峻,王明泰,张代辉,符蓉.理化检验:化学分册),2011,47(5):586-589.
[5] United States Environmental Protection Agency.Section 3 Registration for New Use of Mesotrione on Asparagus,Grass Grown for Seed,Oats,Okra,Rhubarb,Sugarcane and Sorghum.Washington DC:Office of Prevention,Pesticides and Toxic Substances,2007.
[6] Wang M,Duan J S,Sun M N,Zhang Y,Gao T C.Mod.Agrochem.(王梅,段劲生,孙明娜,张勇,高同春.现代农药),2011,10(1):38-40.
[7] Kong D Y,Shi L L,Shan Z J,Wu X W,Wang F,Gao S X.ChinaEnviron.Sci.( 孔德洋,石利利,单正军,吴星卫,王菲,高士祥.中国环境科学),2008,28(8):753-757.
[8] Zhang X,Liu Z H,Yang G Y,Yang L,Duan Y X,Liu C B,Chen Y K,Miao M M.J.Instrum.Anal.(张霞,刘志华,杨光宇,杨柳,段沅杏,刘春波,陈永宽,缪明明.分析测试学报),2014,33(5):545-550.
[9] Zhou C Y,Tang B B,Xi C X,Chen D D,Zhang L,Peng T,Wang G M,Chen Z Q.J.Instrum.Anal.(周春燕,唐柏彬,郗存显,陈冬东,张雷,彭涛,王国民,陈志琼.分析测试学报),2013,32(10):1212-1216.
[10] Alferness P,Wiebe L.J.Agric.FoodChem.,2002,50(14):3926-3934.
[11] Wu Y F,Xu J J,Li Z G,Zhang X C,Yin Y J,Zhang S H,Lin Y Y,Yu W J.Agrochemicals(吴艳芳,徐家俊,李治国,张新昌,尹云吉,张淑红,林妍妍,禹文静.农药),2009,48(3):183-184.
[12] Pang M H,Liu S,Zhang L H,Kang Z H,Tao B,Wu L Q,Liu Y C.J.Agric.Univ.Hebei(庞民好,刘顺,张利辉,康占海,陶晡,吴立强,刘颖超.河北农业大学学报),2007,30(5):75-78.
[13] Durand S,Sancelme M,Besse-Hoggan P,Combourieu B .Chemosphere,2010,81(3):372-380.
[14] Stoob K,Singer H P,Goetz C W,Ruff M,Mueller S R.J.Chromatogr.A,2005,1097(1):138-147.
[15] Gervais G,Brosillon S,Laplanche A,Helen C.J.Chromatogr.A,2008,1202(2):163 -172.
[16] Freitas L G,Götz C W,Ruff M,Singer H P,Müller S R.J.Chromatogr.A,2004,1028(2):277-286.
[17] Barchanska H,Rusek M,Szatkowska A.Environ.Monit.Assess.,2012,184(1):321-334.
[18] Yan F L,Ma J P,Tan P G,Lu X,Jiang L H.J.Instrum.Anal.(闫凤丽,马继平,谭培功,卢曦,姜莲华.分析测试学报),2013,32(11):1328-1332.
[19] Yang Q H,Cheng X Y,Yang P,Qian S,Dan D Z.Chin.J.Anal.Chem.(杨秋红,程小艳,杨坪,钱蜀,但德忠.分析化学),2011,39(8):1208-1212.
[20] Alekseeva T,Kolyagin Y,Sancelme M,Besse-Hoggan P.Chemosphere,2014,111:177-183.
[21] Pinna M V,Roggero P P,Seddaiu G,Pusino A.Chemosphere,2014,111:372-378.
Determination of Trace Amounts of Mesotrione in Water by Solid Phase Extraction and High Performance Liquid Chromatography
WANG Xiao-mei1,2,TAN Pei-gong3,CAO Zheng-mei3,LANG Yin-hai1,2*
(1.College of Environmental Science and Engineering,Ocean University of China,Qingdao 266100,China;2.Key Laboratory of Marine Environment and Ecology,Ministry of Education,Ocean University of China,Qingdao 266100,China;3.Qingdao Environmental Monitoring Centre,Qingdao 266100,China)
A solid phase extraction coupled with high performance liquid chromatographic(HPLC) method was developed for the determination of trace amounts of mesotrione in water.The pH values of the samples were initially adjusted to 3.0-4.0 with phosphoric acid,and then enriched and purified with a C18solid-phase extraction column.The mesotrione fraction was eluted with acetonitrile-water(1∶1) and then concentrated to 10 mL.Mesotrione was determined by HPLC equipped with a Thermo Hypersil-C18column(250 mm×4.6 mm,5 μm),by using a mixture of acetonitrile-phosphoric acid solution(45∶55,pH 3.0) as mobile phase at a constant flow of 0.5 mL/min.The column temperature was set at 30 ℃ and the wavelength of ultraviolet(UV) detection was set at 233 nm.The experimental results showed that a good linear relationship(r>0.999 7) between peak area and mesotrione concentration was obtained in the range of 0.4-20 μg·L-1.The method detection limit for mesotrione was 0.039 μg·L-1.The average recoveries were in the range of 90.3%-94.7% at spiked levels of 1,5,10 μg·L-1with relative standard deviations of 3.5%-4.6%.The method was applied in the determination of trace amounts of mesotrione in surface water and wastewater with recoveries of 89.9%-93.2% at spiked amounts of 5 μg·L-1and 10 μg·L-1.The method showed the advantages of simplicity, rapidness and sensitivity,and was suitable for the determination of trace amounts of mesotrione in water.
mesotrione;solid phase extraction;high performance liquid chromatography(HPLC);acetonitrile;water
2014-10-11;
2014-10-25
国家环境保护标准制修订项目(2012-44)
10.3969/j.issn.1004-4957.2015.02.016
O657.72;S482.4
A
1004-4957(2015)02-0216-05
*通讯作者:郎印海,博士,教授,研究方向:污染控制与生态修复,Tel:0532-66786308,E-mail:yhlang@ouc.edu.cn
猜你喜欢
世界农药(2021年8期)2021-09-02
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
现代农药(2018年1期)2018-03-01
山东化工(2017年12期)2017-09-06
无机化学学报(2016年8期)2016-12-06
化学分析计量(2016年1期)2016-03-14
世界热带农业信息(2014年10期)2014-11-13
现代农业科技(2009年19期)2009-03-20
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
